Amniotic Fluid Embolus (Anaphylactoid Syndrome of Pregnancy)
Renee’ Jones
Steven L. Clark
Amniotic fluid embolus (AFE), also referred to as anaphylactoid syndrome of pregnancy, is an extremely rare and catastrophic event that occurs when amniotic fluid gains entrance into the maternal circulation, causing potentially life-threatening maternal reactions.1 This enigmatic phenomenon is classically characterized by hypoxia, hypotension, cardiovascular collapse, and coagulopathy. It has been documented worldwide, with an estimated incidence of 1 in 8,000 to 1 in 83,000 deliveries.2 The incidence in the United States is estimated between 1 in 8,000 and 1 in 30,000 deliveries.2 A population-based study of over one million deliveries in California during 1994–1995 reported an incidence of 1 in 20,000 deliveries.3 However, it is important to note that the true incidence of AFE is not known, because of the difficulty in confirming the diagnosis and inconsistent reporting of nonfatal cases. The maternal mortality rate from AFE is between 61% and 86%, and makes it a leading cause of death in Western industrialized countries.2 Many women with suspected AFE die within the first hour after the onset of classic symptoms. Only 15% of patients who survive are neurologically intact. The fetal mortality rate is 21%, and, of the neonates who survive, 50% experience neurologic injury.4 AFE continues to be the most lethal maternal complication in obstetrics and remains both unpreventable and unpredictable. Nevertheless, several significant advances have produced a better understanding of this complex condition.
This chapter provides an overview of essential concepts related to AFE. It presents an historical perspective that briefly chronicles early experience with this disorder, and describes The National Registry for Amniotic Fluid Embolus, from which data analysis has yielded valuable information. Issues that provide the framework for care are addressed, including description and etiology of the disorder, pathophysiology, clinical presentation, diagnostic criteria, and clinical management strategies, along with the inherent need for professional collaboration, communication, and teamwork in the clinical setting.
Historical Perspective
The first published case report of AFE is attributed to Meyer in 1926.5 However, the first anatomic description of what may have been an AFE was provided by Baillie in 1789, as he described a case of uterine rupture.6 In 1941, this condition became widely recognized when Steiner and Lushbaugh published their work.7 They described autopsy findings in eight pregnant women with sudden shock and pulmonary edema that occurred during labor. In all cases, they found emboli in small pulmonary vessels that contained fat-positive aggregates and squamous epithelial cells, presumably of fetal origin. This concept is depicted in Figure 19-1 (A and B). Each figure illustrates the presence of squamous cells within the pulmonary artery vasculature at autopsy in women who died from AFE.
In a follow-up report by Liban and Raz in 1969, they described the presence of cellular debris throughout the kidneys, liver, spleen, pancreas, and brain of fourteen women who died from AFE.8 Attwood and Rome published a report in 1975 in which they described the presence of incomplete uterine tears in cases diagnosed as AFE. They postulated that such tears may provide a point of entrance for amniotic fluid or other debris into the maternal system.6 It was not until 1976 that Resnik reported the presence of mucin and fetal squamous cells in blood aspirated from a central venous catheter in a woman who survived a suspected AFE.9 Similar
findings have also been documented in cases where there was no evidence suggestive of AFE. Figure 19-2 illustrates the presence of squamous cells identified in a buffy-coat preparation of blood aspirated from a pulmonary artery catheter in a woman with no signs or symptoms of AFE.
findings have also been documented in cases where there was no evidence suggestive of AFE. Figure 19-2 illustrates the presence of squamous cells identified in a buffy-coat preparation of blood aspirated from a pulmonary artery catheter in a woman with no signs or symptoms of AFE.
 Figure 19-1 (A and B) Squamous cells within the pulmonary artery vasculature at autopsy of a woman who died secondary to AFE. |
Since the initial description of AFE, more than 300 case reports have appeared in the literature.1 Although most cases reportedly occurred during labor, sudden death in pregnancy has been attributed to AFE under a wide variety of circumstances. In 1948, Eastman cautioned that the diagnosis of AFE should not be a wastebasket for cases of unexplained maternal death in labor.10 Subsequent efforts to better understand the syndrome of AFE make such errors less likely today.
The National Registry
A National Registry for Amniotic Fluid Embolus was formed in 1995 by Clark and colleagues.4 Data for the registry were collected between 1983 and 1993 and yielded sixty-nine cases. Data analysis indicated that exposure of the maternal circulation to small amounts of amniotic fluid initiated a syndrome similar to that of anaphylaxis and/or septic shock. Amniotic fluid acts as a foreign substance, similar to that of bacterial endotoxin or a specific antigen, and subsequently stimulates the release of several primary and secondary endogenous mediators. Included in this response is the release of histamine, bradykinin, cytokines, prostaglandins, leukotrienes, thromboxane, and arachidonic acid metabolites into the maternal circulation. It is postulated that the release of these mediators is responsible for development of the severe physiologic sequelae associated with AFE. These include profound hypoxia, myocardial depression, decreased cardiac output, pulmonary hypertension, and disseminated intravascular coagulation (DIC). Based on analysis of data from the registry, the authors suggested that the term “amniotic fluid embolism” be discarded and the syndrome of acute peripartum hypoxia, hemodynamic collapse, and coagulopathy should be designated in a more descriptive manner, as “anaphylactoid syndrome of pregnancy.”4
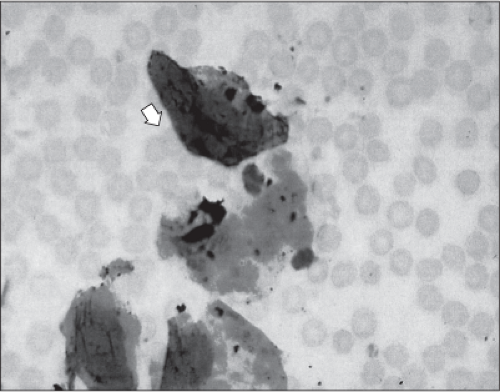 Figure 19-2 Squamous cells identified in a buffy-coat preparation of blood aspirated from a pulmonary artery catheter in a woman with no evidence of compromise. (Photo courtesy of Gary D.V. Hankins, MD.) |
The U.K. Obstetric Surveillance System, in 2005, conducted a population-based cohort study to estimate the incidence of AFE and to investigate risk factors, management, and mortality.2 The findings showed that the risk
of amniotic fluid embolism was greater in induction of labor, multiple pregnancies, and ethnic minority women over 35 years of age. Almost half of the cases of amniotic fluid embolism occurred after delivery, and three fourths of the time, the woman had delivered by Cesarean section.
of amniotic fluid embolism was greater in induction of labor, multiple pregnancies, and ethnic minority women over 35 years of age. Almost half of the cases of amniotic fluid embolism occurred after delivery, and three fourths of the time, the woman had delivered by Cesarean section.
Description and Etiology
Amniotic fluid is made up of maternal extracellular fluid, fetal urine, fetal squamous cells, lanugo, vernix caseosa, mucin, meconium, arachidonic acid metabolites, and, late in pregnancy, increased concentrations of prostaglandins.11 This fluid and the surrounding sac provide an important protective mechanism for the developing fetus. Prior to labor, amniotic fluid does not normally enter the maternal circulation, because it is sealed within the amniotic sac. It has been postulated that amniotic fluid may enter the maternal circulation in one of three ways: 1) through the endocervix following rupture of amniotic membranes; 2) at the site of placental separation; or 3) at the site of uterine trauma, often in the form of lacerations that occur during the course of normal labor, fetal descent, and delivery.12 In addition, introduction of amniotic fluid may also result from placental abruption and the accompanying clinical or sub-clinical disruption of fetal membranes. Once this barrier or membrane is ruptured, alteration of the pressure gradient may allow amniotic fluid to enter the uterine vessels and maternal venous circulation.13 Another potential site for amniotic fluid entry is through small tears in the lower uterine segment and endocervical vessels.11
Early anecdotal reports suggested a possible causal relationship between hypertonic uterine contractions or administration of oxytocin and AFE. The historical anecdotal association between hypertonic uterine contractions and the onset of symptoms of AFE was addressed by analysis of data from the National Registry.4 These data demonstrated that the hypertonic contractions commonly seen in association with AFE appear to be a result of the release of catecholamines into the circulation and part of the initial human hemodynamic response to any massive physiologic insult.4 Under such circumstances, norepinephrine, in particular, acts as a potent uterotonic agent.4,14 While the association of hypertonic uterine contractions and AFE appears to be valid, it is the physiologic response to AFE that causes the hypertonic contractions rather than the converse. In fact, there is complete cessation of uterine blood flow in the presence of even moderate contractions; thus, a hypertonic contraction is the least likely time during the labor process for any exchange between maternal and fetal compartments.1,15 Analysis of National Registry data also revealed that oxytocin was not used with greater frequency in patients who suffered AFE compared with the general population, nor did oxytocin-induced uterine hyperstimulation commonly precede AFE.4 Thus, a causal relationship has been refuted both on statistical and physiologic bases.
Pathophysiology
The pathophysiology of AFE remains incompletely understood. Given the fact that almost universal exchange between maternal and fetal compartments occurs around the time of birth, it is unclear why specific antigens within amniotic fluid, when introduced into the maternal circulation prior to birth, cause an intense pathophysiologic reaction in some women but are benign for most pregnant women. Further, there are no reliable risk factors or warning signs that predict or prevent this catastrophic event. It has been suggested that the amount of fetal debris or the specific type of debris may be significant variables responsible for the anaphylactoid maternal reaction to amniotic fluid.1 Arachidonic acid metabolites have been implicated in the inflammatory response involved in sepsis and anaphylaxis and may be at least partly responsible for similar responses in cases of AFE.16 The ability of arachidonic acid metabolites to cause similar physiologic and hemodynamic changes in animal models as those observed in human AFE has been noted.16 Further, in the animal model of AFE, pretreatment with an inhibitor of leukotriene synthesis has been shown to prevent death.11 There are several clinical findings frequently seen in sepsis and anaphylaxis that are also present in cases of AFE. These include coagulopathy, DIC, left-ventricular failure, and hemodynamic compromise. Clearly, the clinical manifestations of AFE are not identical; fever is unique to septic shock, and cutaneous manifestations are more common in anaphylaxis. Nonetheless, marked similarities exist, which suggest similar pathophysiologic mechanisms. This concept is depicted in Figure 19-3.4 A detailed description of pathophysiologic concepts related to sepsis and septic shock is presented in Chapter 18 of this text. The presence of metabolites symbolizes a humoral mechanism.17 These pathways invoke a proinflammatory response with subsequent release of cytokines and arachidonic acid in AFE.18 This complex inflammatory cascade with mediator release leads to a systemic inflammatory response and subsequently multiple organ system dysfunction or failure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


