Adrenal Gland
Holly L. Williams
Holly L. Williams: Division of Pediatrics, Kaiser, Oakland, California, University of California, San Francisco 94143.
ANATOMY
The bilateral adrenal glands are triangular-shaped dark-yellow structures located high in the retroperitoneum just superior and medial to the upper poles of the kidneys. They weigh 3 to 6 g each, depending on patient age. There are two major divisions of the adrenal gland: The outer portion is the adrenal cortex, and the inner portion is the medulla. The cortex is divided into three separate zones of steroid-producing cells containing abundant smooth endoplasmic reticulum: the outer zona glomerulosa, which produces aldosterone and related mineralocorticoids; and the middle zona fasciculata and the inner zona reticularis, both largely devoted to glucocorticoid and sex hormone production. In the medulla, ectoderm-derived chromaffin cells containing granular endoplasmic reticulum synthesize norepinephrine and epinephrine, which are secreted from vesicles under sympathetic nervous control. The chromaffin cells (pheochromocytes) and the nerve cells account for the two principal childhood medullary neoplasms, pheochromocytoma and neuroblastoma.
The arterial supply of each adrenal gland is derived from superior, middle, and inferior adrenal arteries. The superior adrenal artery is a branch of the inferior phrenic artery that arises from either the aorta or the celiac trunk. The middle adrenal artery arises from the aorta near the origin of the superior mesenteric artery. On the right, the middle adrenal artery runs posterior to the inferior vena cava. Each renal artery gives rise to an inferior adrenal artery. Venous drainage from the glands is largely through a single dominant adrenal vein. The left adrenal vein drains into the left renal vein. On the right, the adrenal vein drains directly into the inferior vena cava posterior to the right lobe of the liver. The right adrenal vein is short and often of large caliber, posing the risk of significant hemorrhage during adrenalectomy (Fig. 42-1).
EMBRYOLOGY AND FETAL ADRENAL FUNCTION
The adrenal cortex arises from mesodermal elements of the adrenogenital ridge, whereas the medulla arises from neuroectoderm. The primordium of the cortex becomes visible by the fourth gestational week (1). At 6 weeks’ gestation, the fetal zone of the adrenal cortex appears and eventually occupies most of the fetal adrenal gland. The cells of the fetal zone have limited 3β-hydroxysteroid dehydrogenase, an enzyme essential for synthesis of active steroid hormones. The large quantities of estrogens reaching the fetus through the placenta are believed to inhibit the enzyme’s expression. The fetal liver can hydroxylate steroids in the 16α position so maternal estrogens reaching the fetus are rapidly converted to estriol, and thus made inactive. The fetal adrenal cannot synthesize cortisol de novo, but it can convert placental progesterone to cortisol (Fig. 42-2). Limited production of active steroids leads to high levels of adrenocorticotropic hormone (ACTH) in the fetus, which probably explains the hyperplastic nature of the fetal adrenal gland. Once placental estrogens disappear after birth, plasma glucocorticoids levels rise, ACTH levels fall, and regression of the fetal zone occurs. Rapid regression of this zone may contribute to fetal adrenal hemorrhage.
During fetal development, medullary or cortical adrenal tissue may remain in various locations to form rests, usually along the aorta and its branches or along the route of descent of the gonads. Large chromaffin masses near the origin of the inferior mesenteric artery are known as the organs of Zuckerkandl. These ectopic sites have surgical significance when a suspected lesion is not found within the normally positioned adrenal glands.
PHYSIOLOGY
Hypothalamic-Pituitary-Adrenal Axis
The principal role of the adrenal glands is the regulation of homeostasis. For this purpose, three types of hormones are synthesized: glucocorticoids, mineralocorticoids, and sex hormones. Control of adrenal hormone synthesis and release is through a series of feedback mechanisms referred to as the hypothalamic-pituitary-adrenal axis (Fig. 42-3). The paraventricular nucleus of the hypothalamus synthesizes corticotrophin-releasing hormone (CRH), which is transported through the pituitary portal system to the adenohypophysis of the anterior pituitary gland. There CRH stimulates the release of ACTH, which increases the production of all three types of adrenal hormones. Release of CRH is under neural control. The normal diurnal variation of CRH release accounts for the cyclical variation in ACTH and therefore cortisol levels. Typically, these peptides are at peak concentrations in the blood shortly before or at the time of awakening, with a gradual decline during the ensuing 24-hour period. Psychological and physical stress lead to enhanced ACTH release (2). Release of CRH is inhibited by cortisol through a negative feedback loop. There is also evidence for negative feedback by ACTH on CRH release, although this appears physiologically subordinate to cortisol feedback inhibition. The complex interactions of these regulatory loops results in fine homeostatic control of plasma cortisol levels.
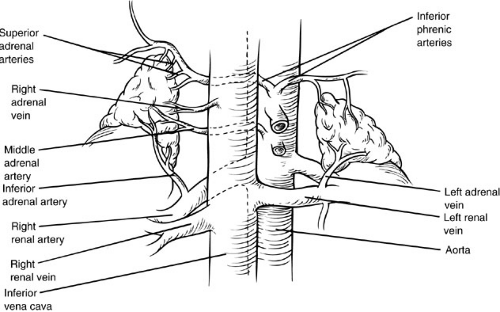 FIGURE 42-1. The arterial and venous anatomy of both adrenal glands. The right adrenal vein is short and drains directly into the inferior vena cava, an important surgical consideration. |
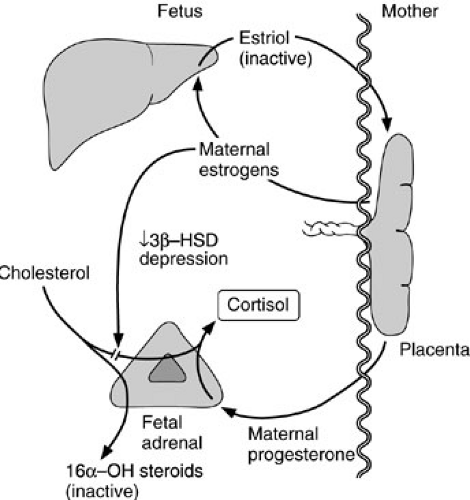 FIGURE 42-2. The fetoplacental unit. Maternal estrogens block de novo cortisol synthesis by the fetal adrenal gland and are converted to estriol by the fetal liver. The fetus synthesizes cortisol by metabolism of maternal progesterone. 3β-HSD, 3β-hydroxysteroid dehydrogenase. |
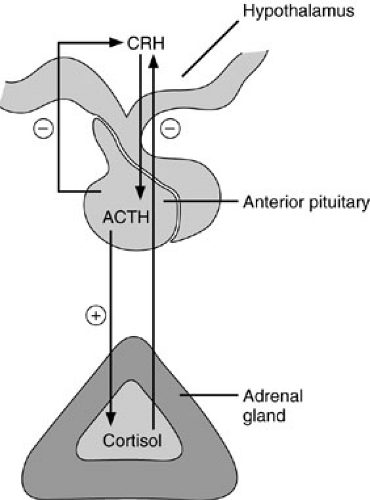 FIGURE 42-3. The hypothalamic-pituitary-adrenal axis. Both the adrenal gland and the pituitary have mechanisms of feedback inhibition of corticotropin-releasing hormone (CRH) release from the hypothalamus. ACTH, adrenocorticotropic hormone. |
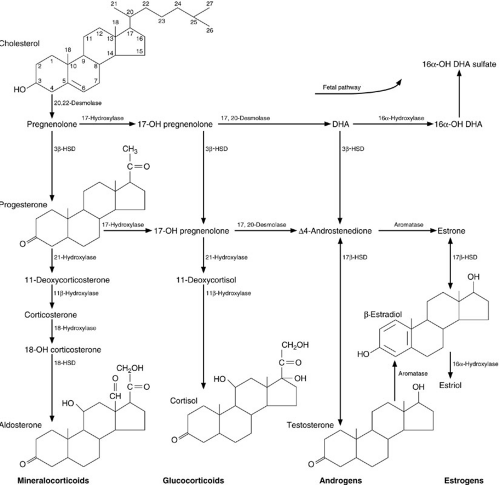 FIGURE 42-4. Intermediary metabolism of adrenal steroids. Most of the enzymatic steps are under the control of adrenocorticotropic hormone. 3β-Hydroxysteroid dehydrogenase (3β-HSD) activity and thereby synthesis of active steroids is inhibited in the fetus by maternal estrogens. 21-Hydroxylase deficiency results in impaired cortisol synthesis, increased ACTH production, and congenital adrenal hyperplasia. |
Glucocorticoid Synthesis and Function
The biosynthesis of steroid hormones begins with cholesterol as the substrate (Fig. 42-4). The initial event, removal of the side chain from position C-20, is a rate-limiting step and is under the control of ACTH. Additional intermediate enzymatic steps are also under ACTH control so overall synthesis of glucocorticoids is regulated by ACTH.
Cortisol is the major glucocorticoid synthesized and is bound by two plasma proteins, albumin (6%) and cortisol-binding protein (90%). The remainder is free in equilibrium with the bound fraction. Unbound steroid molecules cross the plasma membrane of target cells, and are bound by specific cytosolic steroid receptors and transported to the cell nucleus to regulate the synthesis of new messenger RNA and thus the cell protein product. Different target cells have different steroid receptors with variable functions and affinities. Metabolized glucocorticoids are excreted in the urine primarily as 17-hydroxycorticosteroids. Some glucocorticoids undergo cleavage of side chains in the liver, resulting in 17-ketosteroids, which are also eliminated in the urine. A small fraction of free cortisol is also excreted in the urine.
Cortisol has a number of effects on intermediary metabolism, most of which are considered catabolic and oppose the effects of insulin. Glucocorticoids promote proteolysis to provide necessary substrate for gluconeogenesis. Cortisol-induced protein breakdown releases branched-chain and other amino acids, as well as lactate, from muscle. Cortisol induces gluconeogenic enzymes in the liver. Cortisol further promotes hyperglycemia by reducing the utilization of glucose in peripheral tissues; insulin binding by insulin-sensitive tissues and glucose uptake into fat cells are both decreased. An additional catabolic effect of cortisol is the enhancement of lipolysis. Serum-free fatty acids and triglycerides increase under the influence of cortisol. The characteristic truncal obesity of Cushing syndrome (hypercortisolism) may be explained by the fact that truncal adipocytes may be more influenced by insulin (lipogenesis), whereas extremity adipocytes may be more sensitive to cortisol (lipolysis).
Cortisol also has a number of effects on other tissues relevant to the surgeon. Cortisol has both inotropic and chronotropic effects on the myocardium, as well as the ability to increase peripheral vascular resistance. These effects, along with the increase in sodium reabsorption in the distal renal tubule, account for the hypertension frequently seen with cortisol excess. In the gastrointestinal tract, glucocorticoids decrease mucosal cell replication and prostaglandin synthesis. These effects may play a role in the development of the peptic ulcers seen with steroid administration and may also explain the more frequent incidence of acute pancreatitis in steroid-treated patients. Cortisol also has an effect on bone, leading to osteopenia as a result of decreased bone formation. This is believed to be due to decreased osteoblast development, which leads to a deficiency of protein in the extracellular matrix.
Every surgeon is aware of the ability of cortisol to decrease inflammation, blunt the immune response, and impair wound healing. Cortisol excess appears to act principally by decreasing lymphocyte response to specific antigenic stimulation. This explains the efficacy of glucocorticoids in the prevention of rejection in organ transplantation. This blunted response is responsible, at least in part, for the clinical observation that steroid-treated patients are vulnerable to a variety of opportunistic and conventional infectious organisms. Leukocytosis occurs in circulating blood as a result of cortisol administration, but both chemotaxis and phagocytosis are impaired and granulocyte-dependent bacterial killing is less efficient. Wounds have decreased tensile strength in the presence of excess cortisol because both lymphocytes and granulocytes are essential early components of the wound-healing process. In addition, both epithelialization and scar contraction are impaired by cortisol.
Sex Hormone Synthesis and Function
Adrenal androgens (e.g., dehydroepiandrosterone and Δ4-androstenedione) are biologically weak androgens that can be converted by peripheral tissues into more active hormones such as testosterone and dihydrotestosterone. The secretion of adrenal androgens is controlled by ACTH and occurs in synchrony with cortisol in a cyclical manner. Another pituitary regulatory factor named cortical androgen-stimulating hormone appears to stimulate androgen secretion separate from cortisol (3). Prolactin may also play a role in the regulation of adrenal androgen production. Adrenal androgens are synthesized principally in the zona reticularis, the inner layer of the adrenal cortex. This zone is identifiable by 5 years of age and becomes biosynthetically active by 8 years of age. The liver metabolizes androgens, and the metabolites are excreted in the urine as 17-ketosteroids.
The human adrenal gland produces minimal amounts of estrogens. Estrogens may be formed from adrenal androgens by peripheral conversion. Both muscle and adipose tissue can synthesize estrone from Δ4-androstenedione.
Masculinizing effects of androgens include deepening of the voice, deposition of protein in muscle, development of male hair distribution including facial hair, and coarsening of the skin. Estrogens have the opposite effects and enhance breast tissue development. Fetal exposure to androgens leads to wolffian duct development, elongation of the phallic tubercle, and fusion of the labioscrotal folds. The urethra migrates to the tip of the phallic tubercle under the influence of androgens. In the normal female fetus, neither the ovary nor the adrenal glands secrete androgens, and the labial folds, phallic tubercle, and urethra remain in the normal female positions. If the female fetus is exposed to androgens, as in congenital adrenal hyperplasia, the newborn may present with ambiguous or masculinized genitalia. Excess androgen in the male fetus becomes manifest after birth as precocious puberty.
Mineralocorticoid Synthesis and Function
The zona glomerulosa is the principal site of aldosterone synthesis. Although synthetic intermediates such as deoxycorticosterone, corticosterone, and 18-hydroxycorticosterone are produced in the other cortical zones and their production is regulated by ACTH, overall production and release of aldosterone is only weakly regulated by ACTH. The major control mechanism for mineralocorticoid release is the renin-angiotensin system.
Aldosterone circulates free in plasma (37%) and is bound to corticosteroid-binding globulin (22%) and to albumin (41%). A small fraction of aldosterone is excreted unchanged in the urine. The major site for metabolism of aldosterone is in the liver, where the A ring is reduced to form tetrahydroaldosterone and is subsequently conjugated with glucuronic acid. Therefore, patients with impaired hepatocellular function are predictably subject to clinical problems derived from inadequate metabolic processing, leading to mineralocorticoid excess.
Aldosterone affects sodium, potassium, and hydrogen transport in the distal renal tubule. The major action of aldosterone is to increase distal tubular sodium reabsorption while decreasing the reabsorption of potassium, promoting kaliuresis. Increased sodium reabsorption is also associated with increased hydrogen ion excretion in the urine.
Renin-Angiotensin System
Primary physiologic control of mineralocorticoid synthesis and secretion resides in the renin-angiotensin system (4). Decreases in blood pressure and serum sodium concentration lead to release of renin from the macula densa of the juxtaglomerular apparatus in the kidney. Specific prostaglandins and the sympathetic nervous system also regulate renin release, but to a lesser degree. Renin is an enzyme that catalyzes conversion of angiotensinogen to angiotensin I. Angiotensinogen is a relatively large peptide synthesized in the liver. Angiotensin I is in turn processed to angiotensin II by angiotensin-converting enzyme found principally in the lung (Fig. 42-5). Conversion of angiotensin I to angiotensin II is 90% complete in a single pass of blood through the lung.
Angiotensin II acts directly on the adrenocortical cells of the zona glomerulosa by way of a specific cellular receptor to stimulate synthesis of aldosterone. Angiotensin II also acts directly on arterial vessels to increase peripheral vascular resistance and therefore blood pressure. Calcium has a permissive effect on aldosterone synthesis through a mechanism involving desmolase and the conversion of corticosterone to aldosterone. Hyperkalemia also increases aldosterone synthesis, both by direct effect on the cells of the zona glomerulosa and by renin release by the juxtaglomerular cells, thereby increasing urinary excretion of potassium.
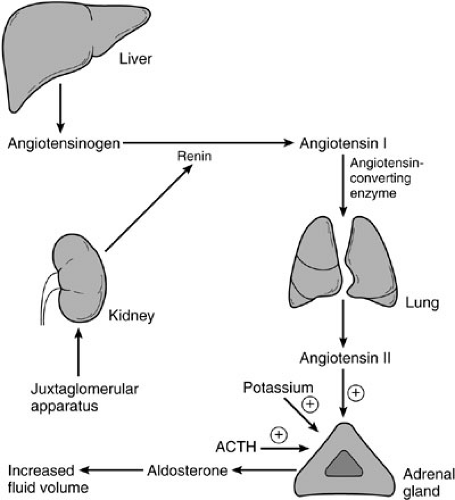 FIGURE 42-5. The renin-angiotensin system. ACTH, adrenocorticotropic hormone. |
In summary, a decrease in blood pressure or in sodium concentration in the renal tubule leads to increased renin secretion and subsequently increased aldosterone secretion. Conversely, sodium loading, intravascular volume excess, and assumption of the supine position result in diminished renin and angiotensin production with decreased aldosterone production. Hyperkalemia increases aldosterone production, thereby promoting potassium excretion in the urine. Likewise, hypokalemia decreases the output of aldosterone and thus provides for potassium conservation in the renal tubule.
Adrenocortical Insufficiency (Addison Disease)
Insufficient production of steroid hormones from the adrenal glands can lead to a potentially life-threatening condition known as Addison disease. The clinical problem can be associated with glucocorticoid, mineralocorticoid insufficiency, or both, depending on the specific cause. The adrenal gland deficiency can be present at birth, secondary to congenital hypoplasia or to an inborn error of steroid metabolism. One form of congenital adrenal hypoplasia is an X-linked disorder in boys, characterized by abnormal adrenal glands with persistence of the fetal zone architecture. The most common inborn errors of steroid metabolism are characterized by defects in glucocorticoid
synthesis and are referred to collectively as congenital adrenal hyperplasia (CAH), described later.
synthesis and are referred to collectively as congenital adrenal hyperplasia (CAH), described later.
Other metabolic defects can specifically affect production of androgens (Noonan syndrome) or mineralocorticoids (hypoaldosteronism). Certain conditions can lead to adrenal insufficiency by destruction of the adrenal glands. These include trauma, infections, adrenoleukodystrophy, and autoimmune diseases. Traumatic delivery or overwhelming infection in the newborn can cause acute hemorrhage of the adrenal glands, resulting in acute adrenal insufficiency. In older children, postsplenectomy sepsis with pneumococcal organisms, meningococcemia, and other causes of overwhelming sepsis can similarly lead to bilateral adrenal hemorrhages and acute adrenal insufficiency, a condition known as the Waterhouse-Friderichsen syndrome. In surgical practice, however, the most common cause of acute adrenal insufficiency is abrupt cessation of long-term exogenous glucocorticoids. Functional adrenal recovery after long-term exogenous steroid therapy may require 6 months or more before restoration of normal hypothalamic-pituitary regulation.
Children with Addison disease have a variety of symptoms, including weakness, fatigue, anorexia, weight loss, vomiting, diarrhea, and abdominal pain. Generally, there is overproduction of ACTH, which stimulates melanocytes resulting in hyperpigmentation. The development of symptoms can be acute or chronic, but if the diagnosis is not made in a timely fashion, adrenal crisis can ensue with resulting hypotension and hypoglycemia causing seizures. Laboratory tests reveal low serum sodium and chloride levels, whereas the potassium concentration can be elevated. Fasting glucose levels can be low, and the fasting morning cortisol level is usually low, with an elevated plasma ACTH. An ACTH stimulation test shows little or no rise in the plasma cortisol levels because the end organ, the adrenal gland, has failed or is unresponsive to stimulation.
Treatment of Addison disease is conceptually simple. Deficient steroid hormones must be replaced. A glucocorticoid, such as hydrocortisone in infants and prednisone in older children, is provided along with a mineralocorticoid, such as fludrocortisone. Doses of glucocorticoids need to be increased and often given parenterally during periods of stress or crisis, as in the setting of surgery.
Adrenal Medullary Function
Chromaffin cells of the adrenal medulla contain catecholamines—dopamine, epinephrine, and norepinephrine— stored in cytoplasmic granules. Release of these catecholamines is under the control of preganglionic sympathetic nerve endings, which secrete acetylcholine. Acetylcholine triggers calcium-dependent exocytosis of the cytoplasmic storage granules, resulting in catecholamine release into the circulation. In essence, the adrenal medulla functions as a sympathetic nerve ganglion. Stimulation of the adrenal nerves causes rapid release of medullary hormones with affecting cortical activity.
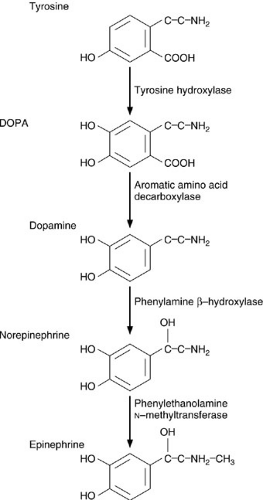 FIGURE 42-6. Intermediary metabolism of catecholamines in the adrenal medulla. DOPA, dihydroxyphenylalanine. |
The catecholamine synthetic pathway is outlined in Fig. 42-6. The process begins with tyrosine, a nonessential amino acid derived from the essential amino acid, phenylalanine. Tyrosine is converted into dihydroxyphenylalanine (DOPA) by tyrosine hydroxylase. This rate-limiting step in the catecholamine synthetic pathway is regulated by acetylcholine. DOPA decarboxylase then converts DOPA into dopamine, which is converted to norepinephrine by phenylamine β-hydroxylase. The final step is conversion of norepinephrine to epinephrine by phenylethylamine N-methyltransferase.
A portion of the released norepinephrine and epinephrine is taken up by the chromaffin cells, and the remainder enters the general circulation. The catecholamines are metabolized by neurons and by enzymatic degradation
in the liver, gut, and kidney. Catecholamines taken up by neurons are metabolized by monamine oxidase to yield vanillylmandelic acid. Carboxyl-o-methyl transferase acts on the compounds at nonneuronal sites to yield normetanephrine from norepinephrine and metanephrine from epinephrine. A fraction of the catecholamines and their metabolites are excreted in the urine, and this can be helpful in diagnosing pheochromocytomas.
in the liver, gut, and kidney. Catecholamines taken up by neurons are metabolized by monamine oxidase to yield vanillylmandelic acid. Carboxyl-o-methyl transferase acts on the compounds at nonneuronal sites to yield normetanephrine from norepinephrine and metanephrine from epinephrine. A fraction of the catecholamines and their metabolites are excreted in the urine, and this can be helpful in diagnosing pheochromocytomas.
Catecholamines are released normally into the circulation from the adrenal gland in a ratio of about 80% epinephrine to 20% norepinephrine. About one-half of plasma catecholamines are loosely bound to protein. The half-life of free epinephrine and norepinephrine is less than 1 minute. Basal levels of epinephrine in plasma range from 20 to 50 pg per mL, but vary considerably depending on the environment and recent stimuli. Transient rises in plasma levels predictably occur with pain and anxiety. Increases in circulating catecholamines also occur with upright posture, hypoglycemia, cold, and cigarette smoking. Two primary feedback inhibitory mechanisms regulate adrenal medullary catecholamine release. First, norepinephrine acts directly on presynaptic, preganglionic α2-recep-tors to decrease the release of acetylcholine. Second, high concentrations of norepinephrine inhibit tyrosine hydroxylase activity, thus inhibiting synthesis of norepinephrine.
Unlike steroids, catecholamines act through cell-surface receptors and target cells do not require the uptake of the catecholamine ligand to respond. All catecholamine effects occur rapidly when compared with the effects of steroid hormones. Although receptors for catecholamines exist in many tissues, the initial biochemical characterization of the receptors was done in smooth muscle cells. α-Receptors lead to smooth muscle contraction, whereas β-receptors lead to relaxation. These two groups of receptors were later subdivided into α1– and α2-receptors and β1– and β2-receptors. α1-Receptors lead predominantly to smooth muscle contraction in peripheral vascular beds and in the uterus. α2-Receptors suppress the release of norepinephrine and acetylcholine from presynaptic terminals and mediate platelet aggregation. β1-Receptors increase inotropy and chronotropy in cardiac muscle, induce lipolysis in adipocytes, and decrease use of glucose in most cells. β2-Receptors lead to smooth muscle relaxation in the conducting airways. Epinephrine and norepinephrine both have α-agonist properties, but the effects are tissue and dose specific. Epinephrine is also a β-agonist.
Other mechanisms can modulate the physiologic effects of catecholamines. Like most ligand-receptor systems, catecholamines downregulate receptors in the presence of hormone excess; similarly, receptors are upregulated in the absence of the hormone. This helps explain the phenomenon of tachyphylaxis to high doses of exogenous catecholamines and also hypersensitivity to catecholamines after surgical sympathectomy. Thyroxine also enhances catecholamine effects by increasing intracellular levels of cyclic adenosine monophosphate (cAMP) in receptor cells. Because cAMP is part of the catecholamine signal transduction process, these target cells have increased sensitivity to catecholamines for a given number of receptors.
PATHOLOGY OF HORMONE OVERPRODUCTION
Most surgeons concerned with the adrenal gland deal with conditions of hormone overproduction, often as the result of tumors. In addition, the pediatric surgeon must be knowledgeable about conditions of hormone overproduction due to inborn errors of metabolism, such as congenital adrenal hyperplasia.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia, also known as the adrenogenital syndrome in girls, is associated with inherited enzymatic defects in steroid synthesis. These defects interfere with the production of normal amounts of cortisol and, by loss of negative feedback, lead to enhanced ACTH production. The increased ACTH levels act early in the steroid biosynthetic pathway to yield exaggerated levels of androgenic intermediaries. Thus, girls are born with masculinized or ambiguous genitalia, a form of pseudohermaphroditism. The degree of masculinization of the external genitalia is related to the timing and amount of androgen production in the fetus. The spectrum of anomalies ranges from mild clitoromegaly, with a normal-appearing vagina, to a malelike phallus, with a urethra extending to the apex and no perineal vaginal orifice. Usually, the disorder falls between these extremes, with clitoral enlargement, labioscrotal fusion, and a urogenital sinus opening at the base of the clitoris. The internal female organs—uterus, fallopian tubes, ovaries, and upper vagina—are normal (5). If mineralocorticoid deficiency is also present, the syndrome may be associated with inability to maintain serum sodium and intravascular volume levels. This is referred to as salt wasting. An infant boy has only increased scrotal pigmentation from the ACTH-stimulated melanocytes, and CAH is usually not diagnosed until adrenal crisis develops 1 to 2 weeks after birth or when he experiences precocious puberty.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


