Acute Respiratory Disorders
Jeffrey A. Whitsett
Ward R. Rice
Gloria S. Pryhuber
Susan E. Wert
Successful adaptation to air breathing at the time of birth is the culmination of an orderly process of pulmonary cell growth and differentiation, leading to alveolar and capillary surfaces capable of providing oxygen and eliminating carbon dioxide. Failure to achieve adequate gas exchange at birth represents a major cause of perinatal morbidity and mortality. This chapter reviews the common disorders of neonatal respiratory adaptation, including respiratory distress syndrome (RDS), pulmonary meconium aspiration syndrome (MAS), pulmonary hypertension, pneumonia, air leak, pulmonary hemorrhage, transient tachypnea of the newborn (TTN), and other causes of acute respiratory dysfunction in the perinatal period. The clinical manifestations and therapy of these disorders are discussed in the context of the morphologic, biochemical, and physiologic factors critical to normal pulmonary growth, maturation, and function in the newborn.
▪ HUMAN LUNG DEVELOPMENT
Human lung development is normally divided into five distinct stages of organogenesis, which describe the histologic changes that the lung undergoes during morphogenesis and maturation of its structural elements (1,2). These five stages include the embryonic, pseudoglandular, canalicular, saccular, and alveolar stages of lung development. Human lung development is initiated during the early embryonic period of gestation (3 to 7 weeks postcoitum) as a small saccular outgrowth of the ventral foregut called the respiratory diverticulum. During the subsequent pseudoglandular stage of lung development (5 to 17 weeks postcoitum), formation of the conducting airways, that is, the tracheobronchial tree, occurs by elongation and repetitive branching of the primitive bronchial tubules. Vascularization of the surrounding mesenchyme with formation of the air-blood barrier, that is, the alveolar-capillary membrane, occurs during the canalicular stage of lung development (16 to 26 weeks postcoitum). Cytodifferentiation of bronchiolar and alveolar epithelial cells is also initiated during this stage. Enlargement and expansion of the peripheral air spaces occur during the saccular stage of lung development (24 to 38 weeks postcoitum), resulting in the formation of primitive sac-like alveoli and thick interalveolar septa. Formation of thin secondary alveolar septa and remodeling of the capillary bed occur during the alveolar stage of lung development (36 weeks postcoitum to 8 years of age), giving rise to the mature alveolar organization of the adult lung (Fig. 26.1).
The human lung is a derivative of the primitive foregut endoderm and the surrounding splanchnic mesoderm (3). The primitive respiratory diverticulum appears at 3 weeks of gestation as an enlargement of the caudal end of the laryngotracheal sulcus located in the median pharyngeal groove, which is an outgrowth of the ventral wall of the primitive esophagus. During the 4th week of gestation, the respiratory diverticulum enlarges and subdivides into the left and right primary bronchi (see Fig. 26.1A and B). The primitive lung grows caudally, expanding into the mesenchyme surrounding the primitive foregut, while the trachea becomes separated from the esophagus by a band of mesenchymal tissue called the tracheoesophageal septum. Between 4 and 5 weeks of gestation, the left and right primary bronchi subdivide to produce secondary, or lobar, bronchi (see Fig. 26.1C and D). Further subdivision of lobar bronchi into tertiary or segmental bronchi occurs during the 6th week of gestation, with the lung taking on a lobulated appearance as the segmental buds are formed (see Fig. 26.1E and F). The developing respiratory tract is lined by endodermally derived epithelium that forms the conducting airways and alveoli. The surrounding mesoderm is composed of mesenchymal cells that will differentiate into connective tissue components, including blood and lymphatic vessels, fibroblasts, smooth muscle cells, and cartilage.
Preacinar blood vessels first appear at the end of week 4 (4,5). Pulmonary arteries arise from the sixth pair of aortic arches and grow into the mesenchyme, where they accompany the developing airways, segmenting with each bronchial subdivision. Pulmonary veins develop as outgrowths of the left atrium of the heart and subdivide several times before connecting to the pulmonary vascular bed. Intra-acinar vessels develop later, in parallel with alveolar formation.
The pseudoglandular stage of fetal lung development extends from about 5 to 17 weeks of gestation and is marked by the formation of the bronchial portion of the lung. This occurs through a process known as branching morphogenesis (6), during which the segmental tubules of the developing lung undergo repetitive lateral and terminal dichotomous branching to form the primitive bronchial tree (see Fig. 26.1G and H). By week 17 of gestation, the segmental bronchi have subdivided to produce approximately 23 generations of bronchial tubules ending in the terminal bronchioles. These bronchial tubules are lined initially by a pseudostratified columnar epithelium containing large pools of glycogen. A prominent basement membrane underlies the epithelium, and mesenchymal cells adjacent to these tubules differentiate into fibroblasts, which become organized in a circumferential orientation, perpendicular to the long axis of the tubules. As branching progresses, pseudostratified columnar epithelium is reduced to a tall columnar epithelium, especially in distal regions of the bronchial tree. During this period, cytodifferentiation of the airway epithelium occurs in a centrifugal direction with ciliated, nonciliated secretory, goblet, neuroendocrine, and basal cells appearing first in the more proximal airways (7,8). Cartilage, smooth muscle cells, and mucous glands are also found in the trachea during the pseudoglandular stage of development and extend as far as the segmental bronchi.
The canalicular stage of fetal lung development extends from week 16 to 26 of gestation. By the end of week 16, the terminal bronchioles have divided into two or more respiratory bronchioles that have subdivided into small clusters of short acinar tubules and buds lined by cuboidal epithelium. These structures undergo further differentiation and maturation to become the adult respiratory unit, or pulmonary acinus, consisting of the alveolated respiratory bronchiole, alveolar ducts, and alveoli. Clusters of acinar tubules and buds continue to grow by lengthening, subdividing, and widening at the expense of the surrounding mesenchyme (Fig. 26.2A). This peripheral growth is accompanied by the formation of intraacinar capillaries, which align themselves around the air spaces, establishing contact with the overlying cuboidal epithelium. During this stage of lung development, type II epithelial cell differentiation occurs in acinar tubules with formation of intracellular multivesicular bodies and multilamellar bodies, the storage form of pulmonary surfactant phospholipids (9). Type I epithelial cell differentiation occurs in conjunction with development of the airblood barrier, wherever endothelial cells of the developing capillary system come into contact with the overlying epithelial cells.
During the saccular stage of fetal lung development, which extends from week 24 to 38 of gestation, the terminal clusters of acinar tubules and buds begin to dilate and expand into thin, smooth-walled, transitory ducts and saccules that later become the true alveolar ducts and alveoli of the adult (Fig. 26.2B). During this stage, there is a marked reduction in the amount of interstitial tissue. Intersaccular and interductal septa develop, which contain
a delicate network of collagen fibers and the intra-acinar capillary bed. Near the end of this stage, elastin is deposited in regions in which future interalveolar septa will form (10). Increasing amounts of tubular myelin, the secretory form of pulmonary surfactant, are seen in the air spaces.
a delicate network of collagen fibers and the intra-acinar capillary bed. Near the end of this stage, elastin is deposited in regions in which future interalveolar septa will form (10). Increasing amounts of tubular myelin, the secretory form of pulmonary surfactant, are seen in the air spaces.
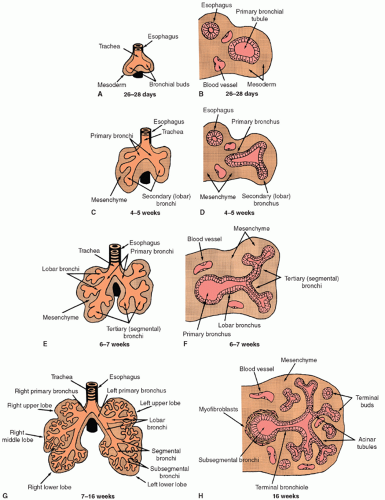 FIGURE 26.1 Lung development during the embryonic (A-F) and pseudoglandular (G and H) stages of organogenesis. The overall branching pattern of the primitive lung (left panels) results in the development of the bronchial tree. The histologic organization of the fetal lung becomes more complex as branching morphogenesis progresses through these stages (right panels). |
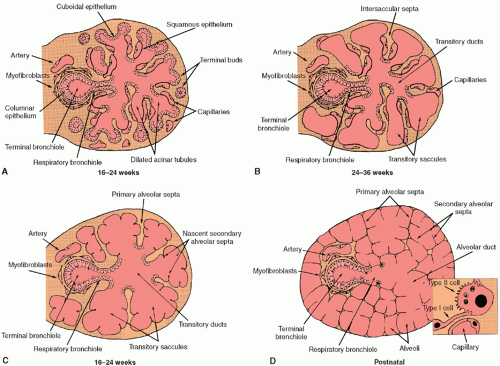 FIGURE 26.2 Lung development during the canalicular (A), saccular (B), and alveolar stages of organogenesis (C and D). Dramatic histologic changes in tissue organization occur during these periods. The adult alveolar epithelium is composed of squamous type I cells and cuboidal type II cells (inset). |
The alveolar stage, which extends from 36 weeks of gestation to between 2 and 8 years of age, is the last stage of lung development and is marked by the formation of secondary alveolar septa, which partition the transitional ducts and saccules into true alveolar ducts and alveoli (Fig. 26.2C and D). This process of alveolarization greatly increases the surface area of the lung available for gas exchange. At the beginning of this period, the secondary interalveolar septa consist of short buds or projections of connective tissue that contain a double capillary network and interstitial cells that are actively synthesizing collagen and elastic fibers. By 5 months of age, these secondary interalveolar septa have lengthened and thinned, and now contain only a single capillary network. Although definitive alveoli can be found in the human lung by 36 weeks of gestation, 85% to 90% of all alveoli are formed within the first 6 months of life (11). Overall the number of alveoli increases by about sixfold between birth and adulthood, that is, from an average of 50 million alveoli in the term lung to 480 million (range: 274 to 790 million) in the adult human lung (12). After the first 6 months of life, alveolar formation occurs at a slower pace until about 2 to 8 years of age, when further growth of the lung becomes proportional to growth of the body (13). The surface area available for gas exchange, and its diffusion capacity, increases linearly with body weight up to about 18 years of age (13). The conducting airways also increase in length and diameter, while airspace and capillary volumes increase coordinately at the expense of interstitial volume.
▪ DEVELOPMENTAL ANOMALIES
Each of these stages of lung development includes distinct changes in tissue organization and cellular differentiation that are important for subsequent growth and maturation of the lung. Structural and functional defects in lung development at birth can often be traced to arrested or aberrant development during one of these periods of organogenesis, often as a result of mutations in genes critical for patterning and growth of the lung, such as the GLI3 gene (Pallister Hall syndrome) (14), which is a component of the Sonic Hedgehog signaling pathway, and the fibroblast growth factor receptor, FGFR2, gene (Pfeiffer, Apert, and Crouzon syndromes) (15,16) that is required for FGF signaling. Lung morphogenesis is determined by complex interactions among cells in the splanchnic mesenchyme and foregut endoderm that are regulated by multiple signaling pathways and transcriptional mechanisms (17). Developmental anomalies of the lung occur through defective division and differentiation of the lung bud, of the left or right bronchial bud or of the trachea and esophagus. Pulmonary agenesis, tracheal and bronchial malformations, tracheoesophageal fistulas, ectopic
lobes, bronchogenic, cysts, and cystic pulmonary adenomatoid malformations (CPAM) arise during the embryonic and pseudoglandular stages of lung development (2). Defects in lung growth and morphogenesis underlie common clinical disorders associated with pulmonary hypoplasia or dysplasia, such as congenital diaphragmatic hernia (CDH) and alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV). Pulmonary hypoplasia can be caused by a reduction of space within the pleural cavity, usually as a consequence with another primary developmental defect, such as CDH or by a reduction in the amount of amniotic fluid following premature rupture of membranes or in association with renal dysgenesis Potter syndrome (18,19). RDS and bronchopulmonary dysplasia (BPD) are associated with premature birth at a time when biochemical functions (e.g., surfactant production) and structural functions (e.g., elasticity) of the lung are still underdeveloped.
lobes, bronchogenic, cysts, and cystic pulmonary adenomatoid malformations (CPAM) arise during the embryonic and pseudoglandular stages of lung development (2). Defects in lung growth and morphogenesis underlie common clinical disorders associated with pulmonary hypoplasia or dysplasia, such as congenital diaphragmatic hernia (CDH) and alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV). Pulmonary hypoplasia can be caused by a reduction of space within the pleural cavity, usually as a consequence with another primary developmental defect, such as CDH or by a reduction in the amount of amniotic fluid following premature rupture of membranes or in association with renal dysgenesis Potter syndrome (18,19). RDS and bronchopulmonary dysplasia (BPD) are associated with premature birth at a time when biochemical functions (e.g., surfactant production) and structural functions (e.g., elasticity) of the lung are still underdeveloped.
Hereditary-Genetic Causes of Acute Respiratory Failure in the Newborn
Mutations in the surfactant protein B (SFTPB), C (SFTPC), ABCA3, and thyroid transcription factor-1 (TTF-1) genes are rare causes of acute respiratory failure in neonates (20,21,22). Hereditary ABCA3 and SP-B deficiency cause respiratory distress, generally in full-term infants, within the first day of life. Respiratory failure progresses despite ventilatory support, surfactant replacement, or extracorporeal membrane oxygenation (ECMO), generally causing death from respiratory failure in the first months of life (23,24). Mutations in both ABCA3 and SFTPB genes are inherited as autosomal recessive traits, resulting in the lack of surfactant in the airways, causing atelectasis and respiratory failure. Hereditary SFTPB and ABCA3 deficiency syndromes are generally lethal, although some infants have been treated by lung transplantation. While most homozygous mutations in ABCA3 are lethal in the neonatal period, several less severe mutations are associated with chronic lung disease in infancy and childhood. Mutations in the SFTPC gene are generally inherited as autosomal dominant and cause both acute and chronic interstitial lung disease in newborns, infants, and adults (25,26). SFTPC-related lung disease is associated with the lack of SP-C protein in alveolar lavage and with folding mutations in the proSP-C protein that accumulates in alveolar type II cells causing cell injury. Hemizygous mutations or deletions in NKX2-1, the gene encoding the transcription factor TTF-1, cause variably penetrant lung disease, hypothyroidism, and central nervous system (CNS) abnormalities, causing acute respiratory disease at birth or chronic respiratory disease in infancy (20). Recent genetic studies of newborn infants with acute respiratory failure caused by ACD/MPV identified mutations in the transcription factor, FOXF1 (Forkhead orthologue F1) (27). At present, definitive diagnosis of these genetic disorders causing respiratory failure in newborn infants is best made by identification of the gene mutations by DNA sequencing.
▪ THE SURFACTANT SYSTEM
The unique physical-chemical boundary between the alveolar gases and the highly solvated molecules at the apical surface of the respiratory epithelium generates a region of high surface tension produced by the unequal distribution of molecular forces among water molecules at an air-liquid interface. Surface-active material at this interface in the alveoli provides surface tension-lowering activity that contributes to the remarkable pressure-volume associations characteristic of the lung. This surface-active material, called surfactant, has been subject to intense study in recent decades (9,28,29).
Deficiency or dysfunction of pulmonary surfactant plays a critical role in the pathogenesis of respiratory diseases in the newborn period. Pulmonary surfactant exists in a variety of physical forms when isolated from the alveolar wash of the lung. These physical forms include lamellated and vesicular forms and highly organized tubular myelin. Tubular myelin is highly surface active and, although composed predominately of phospholipids, its unique structure depends on Ca2+ and lung surfactant proteins A (SP-A), B (SP-B), and D (SP-D). Tubular myelin represents the major extracellular pool of surfactant from which a lipid monolayered/multilayered film is generated to produce an interface between the hydrated cellular surfaces and alveolar gas (Fig. 26.3). Lamellated and vesicular forms of surfactant represent nascent or catabolic forms of surfactant material, respectively; the latter is taken up by type II epithelial cells and recycled. Surfactant proteins A, B, C, and D play important roles in the organization and function of the surfactant complex regulating surfactant homeostasis. Alveolar surfactant concentrations are tightly controlled by a variety of mechanisms that modulate lipid and protein synthesis, storage, secretion, and recycling (30,31).
Composition of Surfactant
Pulmonary surfactant is composed primarily of the phospholipids, phosphatidylcholine (PC), and phosphatidylglycerol (PG) (32,33). These lipid molecules are enriched in dipalmitoyl acyl groups attached to a glycerol backbone that pack tightly and generate low surface pressures. Rapid spreading and stability of pulmonary surfactant are achieved by the interactions of surfactant proteins and phospholipids. Surfactant is synthesized and secreted by type II epithelial cells in the alveolus. Synthesis of PC, surfactant proteins, and lamellar bodies, an intracellular storage form of pulmonary surfactant, increases with advancing gestation. Lamellar bodies are secreted into the lung liquid that contributes to the amniotic fluid. The measurement of amniotic fluid PC, dipalmitoylphosphatidylcholine (DPPC), PG, or the surfactant proteins has provided useful biochemical markers that predict lung maturation and the adequacy of lung function at birth (e.g., lecithin-sphingomyelin [L-S] ratio, lamellar body counts, and PG values). Surfactant function can be assessed by a variety of physical and physiologic tests that measure its ability to reduce surface tension at an air-liquid interface and to spread rapidly during dynamic compression and expansion. The Wilhelmy balance, Langmuir trough, pulsating bubble surfactometer, and a variety of animal models have been used to assess the efficacy of surfactant and surfactant replacements (33,34,35).
Control of Surfactant Synthesis and Secretion
Synthesis of pulmonary surfactant is closely linked to the morphologic and biochemical differentiation of alveolar type II cells in the peripheral respiratory epithelium. Interactions between mesenchymal and epithelial cells, mediated by direct cell-cell contact or by paracrine factors, contribute to the differentiation process (14). Endocrine factors also modulate the differentiation of type II epithelial cells and the synthesis of surfactant components. In vivo and in vitro evidence supports the role of glucocorticoids in the modulation of morphologic differentiation and production of phospholipids and surfactant proteins by the lung (36,37).
Phospholipid Synthesis
PC is produced by type II epithelial cells using extracellular substrate, and the glycogen stores that accumulate in the pretype II cells of the fetal lung. Metabolic pathways producing PC depend on the production of phosphatidic acid and a glycerophosphate backbone; the latter is produced as an intermediate of the glycolytic pathway (32,33). The synthesis of PC involves the deacylation of phosphatidic acid and its reaction with cytidine diphosphocholine (CDP-choline) (32,33).
Disaturated forms of PC may be formed de novo, using disaturated acyl precursors or by remodeling, that is, salvage pathway, of phospholipids by deacylation and reacylation reactions. Production of CDP-choline is critical to PC synthesis and is achieved by phosphorylation of choline and transfer to cytidine triphosphate
in a reaction dependent on choline kinase and choline phosphate cytidylyltransferase. Acyl-transferases, for example, LPCATs, regulate the abundance and specificity of the acyl chain in PC. The activities of many of the enzymes in the synthetic pathway for PC increase with advancing gestation in the lung, generally increasing in the last third of gestation (32,33).
in a reaction dependent on choline kinase and choline phosphate cytidylyltransferase. Acyl-transferases, for example, LPCATs, regulate the abundance and specificity of the acyl chain in PC. The activities of many of the enzymes in the synthetic pathway for PC increase with advancing gestation in the lung, generally increasing in the last third of gestation (32,33).
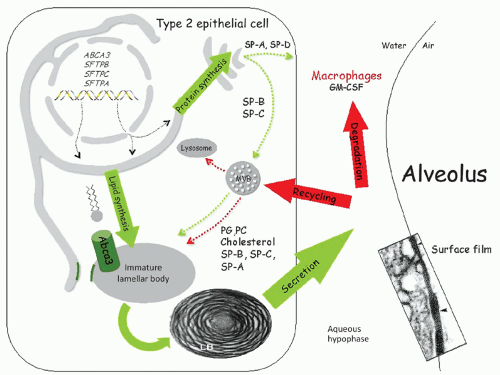 FIGURE 26.3 Surfactant synthesis and metabolism by type II alveolar epithelial cells. The hydrophobic surfactant proteins, SP-B and SP-C, and the phospholipid transporter, ABCA3, are synthesized in the endoplasmic reticulum (ER) and extensively modified during transit through the ER, the Golgi apparatus, and the multivesicular body (MVB) (green dashed arrows). The MVB then fuses with the lamellar body (LB), where final packaging of the surfactant proteins and phospholipids occurs before secretion into the alveolus (green dashed arrows). PC and PG are the most abundant surfactant lipids and are transported into the LB by ABCA3, located in the outer membrane of the LB. The hydrophilic surfactant proteins, SP-A and SP-D, are also synthesized in the ER, glycosylated in the Golgi apparatus, and then secreted into the alveolus (solid green wide arrows). After secretion, surfactant phospholipids and proteins are reorganized into a multilayered, bioactive, surface film at the air-fluid interface, which reduces surface tension in the alveolus. Secreted surfactant phospholipids and proteins are taken up by type II cells and are either catabolized within lysosomes or transported to the LB for recycling (red dashed arrows). A fraction of the secreted surfactant proteins and phospholipids are also taken up and degraded by alveolar macrophages in a process requiring GM-CSF. Diagram courtesy of Timothy Weaver, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH. |
Glucocorticoid Enhancement of Pulmonary Maturation
A variety of hormonal factors influence the rate of production of the enzymes controlling PC synthesis in the developing lung (32,33). Glucocorticoids are the most clinically relevant and useful of these agents (36,37). Studies in fetal lambs and humans demonstrated that administration of glucocorticoid to the dam or mother resulted in precocious respiratory function in prematurely born offspring. The initial clinical studies of Liggins and Howie demonstrated that maternal administration of glucocorticoid decreased the incidence of respiratory distress in premature infants (38). Although the precise mechanisms by which glucocorticoids induce pulmonary maturation and lung function in premature infants have not been discerned, increased PC synthesis and morphologic remodeling of the alveolar architecture, including the thinning of interstitial components of the fetal lung, are observed after glucocorticoid treatment. Glucocorticoids regulate many genes that are associated with the differentiation of the fetal lung, including the genes regulating pulmonary mesenchymal differentiation and those encoding enzymes involved in the synthesis of PC and the surfactant proteins. The effects of glucocorticoid on lung cell differentiation are mediated by glucocorticoid receptors, which, when occupied by hormones, influence gene transcription and mRNA stability, altering the abundance of the proteins synthesized by pulmonary cells. Prenatal glucocorticoid therapy is useful in prevention of RDS in preterm infants.
Other Hormonal Influences
Thyroid hormones that is, T3, T4, thyrotropin-releasing hormone (TRH), estrogens, prolactin, epidermal growth factor, β-adrenergic agents, and other agents that enhance cellular cyclic adenosine monophosphate (cAMP) levels influence pulmonary maturation or biochemical indices of pulmonary maturation (39,40). Both T3 and T4 increase the synthesis of phospholipids in mammalian lung but do not readily cross the placenta.
Surfactant Secretion
Surfactant is stored within type II cells in large lipid-rich organelles called lamellar bodies. Secretion of lamellar bodies occurs by a process of exocytosis that is regulated by a number of physical and hormonal factors. Stretch, the mode of ventilation, and the labor process enhance surfactant secretion and extracellular surfactant pool sizes at birth. Catecholamines and purinoceptor agonists (e.g., adenosine triphosphate) that activate protein kinases enhance phospholipid secretion by type II cells in vitro (33). Recent studies identified the role of a G protein-coupled receptor, GPR116, present on alveolar epithelial cells, that regulates surfactant lipid
pool sizes (41). Hyperglycemia and hyperinsulinemia inhibit surfactant phospholipid secretion (42). Newly secreted surfactant enters the extracellular space and undergoes dramatic structural reorganization to form tubular myelin, a process dependent on SP-A, Ca2+, phospholipids, and SP-B (33). Phospholipids must move from tubular myelin to form monolayers and multilayers at the airliquid interface, thereby reducing collapsing forces in the alveoli.
pool sizes (41). Hyperglycemia and hyperinsulinemia inhibit surfactant phospholipid secretion (42). Newly secreted surfactant enters the extracellular space and undergoes dramatic structural reorganization to form tubular myelin, a process dependent on SP-A, Ca2+, phospholipids, and SP-B (33). Phospholipids must move from tubular myelin to form monolayers and multilayers at the airliquid interface, thereby reducing collapsing forces in the alveoli.
Surfactant Recycling and Catabolism
The process of inflation and deflation produces spent forms of surfactant phospholipids that are taken up by type II cells and reused or catabolized (30,43). Surfactant proteins B and C enhance the reuptake of phospholipids in vitro. In the adult rabbit lung, the half-life of surfactant phospholipids is approximately 6 to 8 hours, and in newborn animals, the half-life is 3.5 days (43). The intracellular and extracellular pools of surfactant are generally larger in the newborn animal than in adults. A relatively small fraction of the alveolar surfactant pool is cleared by catabolism and alveolar macrophages, with most of the surfactant phospholipid recycled or catabolized by type II epithelial cells. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and receptors play a critical role in the regulation of surfactant lipid and protein clearance, acting on the alveolar macrophage. Defects in GM-CSF signaling, caused by autoantibodies to GM-CSF or mutations in its receptors, inhibit alveolar macrophagemediated degradation of surfactant lipids and proteins, leading to their accumulation in the postnatal lung, in turn, causing the syndrome of pulmonary alveolar proteinosis (44). Exogenously administered surfactant phospholipid is reused efficiently by adult and newborn lungs (43). The effects of surfactant replacement therapy are therefore related both to the direct surface tension-lowering properties of surfactant introduced into the airway and to the recycling of the exogenously delivered phospholipids by type II cells.
The Role of Surfactant in Lung Disease
Quantitative and qualitative abnormalities of pulmonary surfactant contribute to the pathogenesis of lung disease in the newborn infant. In premature infants, deficiencies in surfactant production and secretion decrease intracellular and extracellular pools of surfactant, leading to alveolar surfactant insufficiency and atelectasis. Qualitative abnormalities of surfactant are also associated with many types of lung injury. Alveolar-capillary leak, hemorrhage, pulmonary edema, and alveolar cell injury fill the alveolus with proteinaceous material that inactivates surfactant. Serum and nonserum proteins, including albumin, fibrinogen, hemoglobin, and meconium, are potent inactivators of pulmonary surfactant in vivo and in vitro; SP-A, SP-B, and SP-C act synergistically to stabilize the surface properties of phospholipids in the presence of these inactivating proteins. Inhibitory factors associated with surfactant dysfunction in acute lung injury can be overcome by the administration of exogenous surfactants that contain the surfactant proteins.
Surfactant Replacement
The first successful surfactant replacement therapy in humans was reported by Fujiwara et al. in 1980 (45). Natural synthetic and semisynthetic surfactants have been successfully administered into the lungs of premature infants for treatment of RDS, for the treatment of meconium aspiration, and are being tested for therapy of other lung diseases. Surfactant replacement has become standard for prevention and treatment of RDS. Animal surfactant preparations containing phospholipids, SP-B, and SP-C (e.g., Survanta, Curosurf, Infasurf, BLES) and synthetic preparations composed primarily of phospholipids mixed with synthetic surfactant protein-like peptides are in clinical use (35), although the animalbased preparations are most widely used. The surfactant preparations containing surfactant proteins provide highly surface active material to the alveolus. Surfactant replacement also contributes to the pool size of surfactant phospholipids, providing substrate for surfactant synthesis by means of the recycling pathways.
▪ RESPIRATORY DISTRESS SYNDROME
RDS, previously called hyaline membrane disease, is a common cause of morbidity and mortality associated with premature delivery. RDS is a developmental disorder rather than a disease process per se, and it is usually associated with premature birth. The incidence and severity of RDS generally increase with decreasing gestational age at birth and are usually worse in male infants. Infants of diabetic mothers with poor metabolic control and infants born after fetal asphyxia, maternofetal hemorrhage, or after pregnancies complicated by multiple births are at higher risk for RDS. RDS affects approximately 20,000 to 30,000 infants each year in the United States and complicates about 1% of pregnancies. Approximately 50% of the infants born between 26 and 28 weeks of gestation develop RDS, whereas fewer than 20% to 30% of premature infants at 30 to 31 weeks have the disorder.
Clinical Presentation
Infants with RDS present at birth or within several hours after birth with clinical signs of respiratory distress that include tachypnea, grunting, retractions, and cyanosis accompanied by increasing oxygen requirements. Physical findings include rales, poor air exchange, use of accessory muscles of breathing, nasal flaring, and abnormal patterns of respiration that may be complicated by apnea. Chest radiographs are characterized by atelectasis, air bronchograms, and diffuse reticular-granular infiltrates, often progressing to severe bilateral opacity characterized by the term “white-out” (Fig. 26.4). Radiographic patterns in RDS are variable and may not reflect the degree of respiratory compromise.
The infant attempts to maintain alveolar volume by prolonging and increasing expiratory pressures by breathing against a partially closed glottis, causing the grunting noise characteristic of RDS, but often seen in other respiratory disorders as well. Increasing oxygen requirements and the need for ventilatory support often occur
rapidly in the first 24 hours of life and continue for several days thereafter. The clinical course depends on the severity of RDS and the size and maturity of the infant at birth. In uncomplicated RDS, typically seen in more mature infants, recovery occurs over several days, and infants generally no longer require oxygen or ventilatory support after the first week of life. The most premature infants are at greatest risk for severe RDS and frequently develop complications, including CNS hemorrhage, patent ductus arteriosus (PDA), air leak, and infection, which contribute to prolonged requirements for oxygen and ventilatory support.
rapidly in the first 24 hours of life and continue for several days thereafter. The clinical course depends on the severity of RDS and the size and maturity of the infant at birth. In uncomplicated RDS, typically seen in more mature infants, recovery occurs over several days, and infants generally no longer require oxygen or ventilatory support after the first week of life. The most premature infants are at greatest risk for severe RDS and frequently develop complications, including CNS hemorrhage, patent ductus arteriosus (PDA), air leak, and infection, which contribute to prolonged requirements for oxygen and ventilatory support.
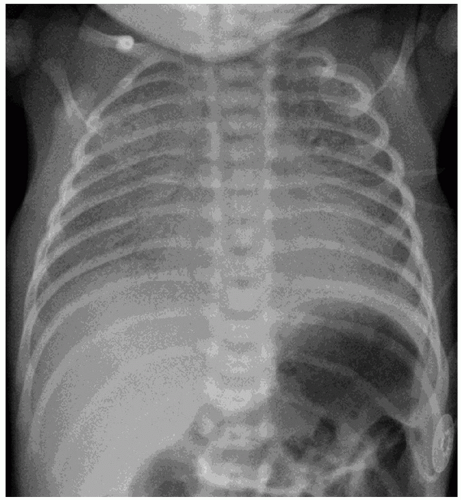 FIGURE 26.4 This premature infant presented with grunting, retractions, and cyanosis after delivery. The diffuse reticular-granular opacification, air bronchograms, and decreased lung volumes in the chest radiograph film indicate RDS. |
Pathology
Pathologic findings early in the course of RDS include atelectasis, pulmonary edema, pulmonary vascular congestion, pulmonary hemorrhage, and evidence of direct injury to the respiratory epithelium (Fig. 26.5). Epithelial cell injury is especially evident in the bronchiolar region of the lung. Histologic findings include the presence of hyaline membranes, the characteristic eosinophilic material derived from bronchial and bronchiolar injury to epithelial cells. Alveolar spaces are generally not inflated, and at autopsy, the lungs of infants with RDS are often airless on passive deflation. Leukocytic infiltration is not observed early in the course of RDS unless complicated by infection. Pulmonary edema, hemorrhage, and hemorrhagic edema are common pathologic features in RDS, especially if the clinical course is further complicated by PDA and congestive heart failure.
Pathophysiology
Avery and Mead first demonstrated the paucity of alveolar surfactant in the lungs of infants dying of RDS (46




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
