Abdominal Wall Defects
Robert K. Minkes
Section of Pediatric Surgery and Department of Surgery, Louisiana University School of Medicine, Pediatric Surgery, Department of Surgery, Children’s Hospital of New Orleans, New Orleans, Louisiana 70118.
Management challenges of infants with abdominal wall defects begin in the prenatal period and can extend many years into the child’s life. The use of prenatal ultrasound and maternal alpha-fetoprotein has led to an increasing number of abdominal wall defects diagnosed before birth. This provides the opportunity to influence neonatal outcome through changes in the management of the pregnancy or delivery, and by prenatal education and counseling of the family (1). Survival of infants with gastroschisis and isolated omphalocele has improved markedly; however, many challenges remain. Our understanding of the normal and abnormal development of the abdominal wall continues to improve and the prenatal and postnatal management of infants with these defects is evolving. Prior to the late 1960s, survival for infants with gastroschisis was rare. Three major advances led to a marked improvement in survival in these babies: parenteral nutrition, the ability to use a silo when primary closure was not possible, and advances in perinatal care in the neonatal intensive care unit (NICU). This chapter reviews the relevant embryology, anatomy, etiology, and management of common abdominal wall defects.
EMBRYOLOGY
The embryology of the anterior abdominal wall and umbilicus is well described (2,3). The details on the molecular and genetic level are less clear. From a descriptive standpoint, the embryo is seen to grow, curl, and fold in on itself. This process represents several integrated events that are specific regarding time and location. Cellular differentiation, proliferation, migration, and deposition of cells occur at different times and rates with respect to the developing embryo and are likely stimulated and regulated by specific growth factors. Several events coincide with the formation of the abdominal wall, and it is not surprising that other anomalies are associated with abdominal wall defects.
The embryo begins as a flat structure within the umbilical ring and is comprised of the epiblast (ectoderm), destined to become neuroectoderm or surface epithelium, and the hypoblast (endoderm), which becomes the inner epithelium of the gut-derived organs. Formation of the third germ layer (mesoblast) occurs with the change in the embryo’s shape. The processes involved to form the mesoblast cell layer include apoptosis of the epithelial basement membrane, phagocytosis of the dead cells and migration of the ectoderm cells from the epithelial layer to the mesodermal layer. Defects in these processes may lead to body stalk abnormalities. As the neuroectoderm and underlying mesoderm proliferate, the embryonic disc is seen to grow and the embryo folds ventrally. During this time, the thoracic and abdominal cavities become distinct from the extraembryonic coelom. The folding of the embryo occurs along two perpendicular axes and involves folding from four directions—the cranial, the caudal, and two lateral aspects of the embryo. The cranial fold contains the embryonic derivatives that will develop into the thoracic and epigastric walls. The hindgut, bladder, and hypogastric wall are derived from the caudal fold. The lateral folds are involved in midgut and lateral abdominal wall development. The ectoderm is continuous with the amnion, and as the embryo elongates, it curls and wraps around, eventually putting the ectoderm-amnion border on the ventral body wall where it forms the umbilical ring around a diminishing yolk sac. The umbilical ring contains the allantois, umbilical vessels, extraembryonic coelom, the vitelline duct, and the associated vitelline vessels.
Following the completion of folding in the fourth week of gestation, the next embryologic event significant in the development of abdominal wall defects is the rapid growth of the midgut from the sixth to the tenth week. During this stage, the midgut elongates at a rate greater than the growth of the abdominal cavity and the bowel herniates
through the umbilical ring into the extraembryonic coelomic space in the umbilical cord. In normal development, the midgut rotates and returns into the abdominal cavity from the tenth to the twelfth week. Without proper abdominal wall development, the viscera remain herniated in the umbilical cord. By the end of the twelfth week the extraembryonic space is obliterated and the intestine return to the abdominal cavity. Similarly, the allantois, vitelline duct, and accompanying vessels are obliterated, leaving one umbilical vein and two umbilical arteries covered by amnion. Defects in these processes result in defects of the abdominal wall.
through the umbilical ring into the extraembryonic coelomic space in the umbilical cord. In normal development, the midgut rotates and returns into the abdominal cavity from the tenth to the twelfth week. Without proper abdominal wall development, the viscera remain herniated in the umbilical cord. By the end of the twelfth week the extraembryonic space is obliterated and the intestine return to the abdominal cavity. Similarly, the allantois, vitelline duct, and accompanying vessels are obliterated, leaving one umbilical vein and two umbilical arteries covered by amnion. Defects in these processes result in defects of the abdominal wall.
PATHOGENESIS OF ABDOMINAL WALL DEFECTS
The pathogenesis of abdominal wall defects is not completely understood and the etiology likely differs depending on the timing of the embryologic abnormality. In general, the earlier the embryologic defect, the more complex the structural anomaly (Table 69-1). Abdominal wall defects may result from failure of mesoderm ingrowth and decreased apoptosis, failure of the umbilical ring to develop, failure of the amnion to approximate with the yolk sac and body stalk, abnormal neuronal differentiation, or nerve ingrowth, and may be under the influence of maternal or environmental factors.
Body stalk anomalies are produced when the embryologic defect occurs in the fourth to sixth week of gestation. Head, spine, and extremity anomalies are observed in addition to the defect of the abdominal wall. Gastroschisis results when the defect occurs in the sixth or seventh week. During normal development, the paired umbilical veins supply the abdominal wall until the omphalomesenteric arteries branch from the aorta and replace them. In the seventh week, the right umbilical vein and the left umbilical artery atrophy. The left umbilical vein and the right omphalomesenteric artery remain to supply the anterior abdominal wall. Gastroschisis (Fig. 69-1) is believed to result from vascular compromise of the right abdominal wall. Ischemia of the right paraumbilical abdominal wall could result from premature atrophy or persistence of the right umbilical vein that may interfere with development of aortic collaterals (4) or a vascular accident of the right omphalomesenteric artery. Ischemia of the right omphalomesenteric artery can produce intestinal atresia, a finding associated with gastroschisis (5). Despite clear differences in the origin of the various abdominal wall defects, there are a small number of fetuses in whom gastroschisis results from prenatal rupture of an omphalocele (6).
TABLE 69-1 Embryology of Abdominal Wall Defects. | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
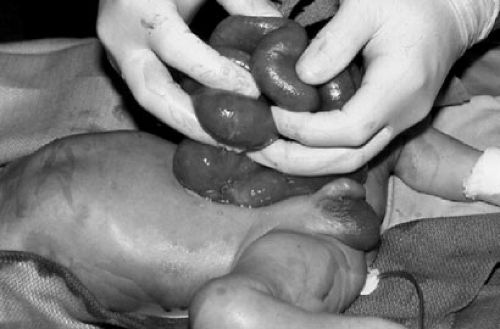 FIGURE 69-1. Gastroschisis demonstrating thickening of eviscerated bowel and lack of normal markings and folds. |
Embryologic anomalies during the seventh and eight weeks can affect multiple areas of the developing abdominal wall and usually involve the cranial or caudal folds
producing a predictable pattern of defects. During this time, the abdominal wall is completed by the fusion of mesodermal myotomes in the midline. The diaphragm and fibrous pericardium are also formed during this time. In addition, the urorectal septum and anterior wall of the bladder and overlying abdominal wall are formed. Defective fusion of the mesodermal myotomes in the upper midline is associated with lack of formation of the septum transversum and failure of the fibrous pericardium to form. The resulting defect termed the pentalogy of Cantrell (Fig. 69-2) is associated with thoracoabdominal ectopia cordis and other cardiac defects, omphalocele, cleft sternum, and diaphragmatic and pericardial defects. The urorectal septum arises between the allantois and hindgut at this time, and separates the cloaca into the urogenital sinus and the anorectal canal. Failure of these structures to form results in cloacal exstrophy: extrophy of the bladder, imperforate anus, omphalocele, and lower neural tube defects. Bladder exstrophy results from failure of the cloacal membrane to develop and defective fusion of the mesodermal myotomes in the lower midline.
producing a predictable pattern of defects. During this time, the abdominal wall is completed by the fusion of mesodermal myotomes in the midline. The diaphragm and fibrous pericardium are also formed during this time. In addition, the urorectal septum and anterior wall of the bladder and overlying abdominal wall are formed. Defective fusion of the mesodermal myotomes in the upper midline is associated with lack of formation of the septum transversum and failure of the fibrous pericardium to form. The resulting defect termed the pentalogy of Cantrell (Fig. 69-2) is associated with thoracoabdominal ectopia cordis and other cardiac defects, omphalocele, cleft sternum, and diaphragmatic and pericardial defects. The urorectal septum arises between the allantois and hindgut at this time, and separates the cloaca into the urogenital sinus and the anorectal canal. Failure of these structures to form results in cloacal exstrophy: extrophy of the bladder, imperforate anus, omphalocele, and lower neural tube defects. Bladder exstrophy results from failure of the cloacal membrane to develop and defective fusion of the mesodermal myotomes in the lower midline.
The most widely accepted mechanism of omphalocele (Fig. 69-3) formation is the improper migration and fusion of the lateral embryonic folds resulting in failure of closure at the umbilical ring. This results in persistent herniation of the midgut. The herniated abdominal viscera are contained within an opaque omphalocele sac. The sac is composed of peritoneum internally, amnion externally, and may contain Wharton’s jelly. The umbilical cord inserts into the sac, and the umbilical vessels radiate onto the wall of the sac. The fascial defect associated with an omphalocele can vary greatly in size. The smallest omphaloceles are sometimes referred to as congenital hernias of the umbilical cord and have a small fascial defect and minimal abdominal contents herniating into the small sac. The larger omphaloceles are more challenging to manage and may have a fascial defect up to 12 cm in diameter and involve almost the entire anterior abdominal wall. Stomach, small bowel, and large bowel often make up the contents of the sac. The liver is found herniating into the sac approximately one-half the time (7). Giant omphaloceles can include the entire gastrointestinal tract, liver, spleen, bladder, and gonads. Because the abdominal viscera develop outside the peritoneal cavity, there is no impetus for the peritoneal cavity to grow and increase in capacity. The small, underdeveloped peritoneal cavity presents a challenge in the management of infants with a giant omphalocele and gastroschisis. Gross’ observation that the abdominal contents had lost its right of domicile to the peritoneal cavity is apropos to these abdominal wall defects.
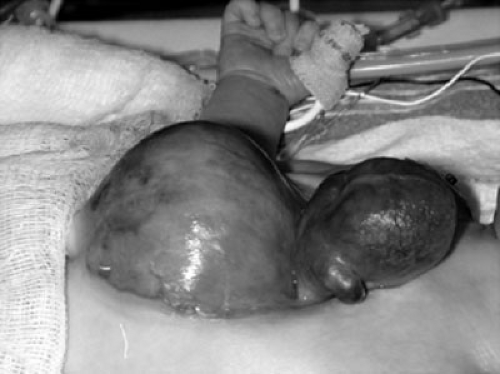 FIGURE 69-2. Pentalogy of Cantrell. Note omphalocele and ectopia cordis. |
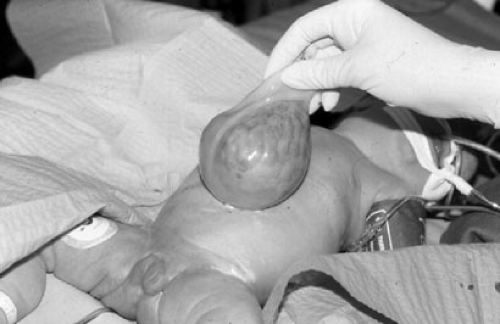 FIGURE 69-3. Appearance of omphalocele. Note umbilical cord originating from omphalocele sac. |
ETIOLOGY
The precipitating cause leading to abdominal wall defects is unclear, but may involve genetic predisposition, environmental, or maternal factors. Socioeconomic factors, reproductive immaturity, medications, illnesses, and nutritional status may contribute. It is postulated that these factors produce placental insufficiency and may also be responsible for the small for gestational age or prematurity that is common among infants with abdominal wall defects. Gastroschisis is associated with an average maternal age younger then 20 years. Folic acid deficiency, hypoxia, and salicylates have been shown to produce abdominal wall defects in laboratory animals, and a more recent population-based study demonstrated periconceptional multivitamin use was associated with a 60% reduction in the risk for nonsyndromic omphalocele (8,9). Several studies suggest that maternal ingestion of certain medications or drugs,
especially those producing vasoconstriction early in pregnancy increases the risk of gastroschisis. Medications that are commonly used for colds, coughs, and pain early in pregnancy may increase both the risk of gastroschisis and small intestine atresia. Among the medications implicated are aspirin, acetaminophen, ibuprofen, pseudoephedrine, and phenylpropanolamine (10,11,12,13). Caution must be used when attributing ingestion of medication as causative because it may have been an underlying illness or a combination of the illness and medication that ultimately produces the defect. There are data to suggest that the risk of abdominal wall defects is increased in conjunction with cigarette smoking (10,11,12,13,14). Marijuana, cocaine, alcohol, and other agents have also been implicated (12,15,16,17,18,19).
especially those producing vasoconstriction early in pregnancy increases the risk of gastroschisis. Medications that are commonly used for colds, coughs, and pain early in pregnancy may increase both the risk of gastroschisis and small intestine atresia. Among the medications implicated are aspirin, acetaminophen, ibuprofen, pseudoephedrine, and phenylpropanolamine (10,11,12,13). Caution must be used when attributing ingestion of medication as causative because it may have been an underlying illness or a combination of the illness and medication that ultimately produces the defect. There are data to suggest that the risk of abdominal wall defects is increased in conjunction with cigarette smoking (10,11,12,13,14). Marijuana, cocaine, alcohol, and other agents have also been implicated (12,15,16,17,18,19).
GENETICS
Familial cases of both omphalocele and gastroschisis have been reported (20). Omphalocele is associated with trisomy 13, 14, 15, 18, and 21 (12) and other genetic syndromes. New syndromes that include omphalocele have more recently been described (21,22). The reports of gastroschisis coinciding with other anomalies such as Hirschsprung’s disease and oromandibular limb hypogenesis suggest that these are sporadic events (23,24). The association of gastroschisis and specific genetic conditions in humans is less well defined. However, gastroschisis can be induced in laboratory animals by both chemicals and radiation (12). In addition, certain strains of mice, such as the HLG inbred mouse, show an increased prevalence of gastroschisis and are more susceptible to radiation injury (25,26). Gastroschisis is also seen in p53-deficient mice exposed to radiation (27). Methods for genome analysis allow for identification of genes that may be responsible for multifactorial or polygenic diseases. A mutation in the bone morphogenic protein gene 1 produces gastroschisis in mice; however, a more recent study found no mutation of this gene in 11 patients with gastroschisis (28). The use of transgenic and knockout animals has begun to shed some light on the complex nature of these defects.
INCIDENCE
The prevalence of congenital abdominal wall defects is 4 to 5 per 10,000 live births. Prenatal ultrasound data indicate an incidence of 1 in 2,500 fetuses. Historically, the incidence of omphalocele has been reported to be 1 in 4,000 births and gastroschisis 1 in 6,000 to 10,000 births. Both gastroschisis and omphalocele occur in males and females equally. More recent reports have indicated that the birth prevalence of gastroschisis is on the increase worldwide, whereas the prevalence of newborns with omphalocele in most regions has not changed or decreased slightly during the same time periods (29). Even where increases in the prevalence of omphalocele have been seen, they are less dramatic than the increases seen for gastroschisis. The reason for the increase is not well defined (29,30,31,32).
PATHOPHYSIOLOGY
Table 69-2 lists the salient differences of these two types of abdominal wall defects. The physiologic impact of an omphalocele is related to its size and, more important, to associated anomalies that can occur in approximately 60% of neonates with an omphalocele. Structural malformations are more common in infants with omphalocele than in those with gastroschisis (33,34,35,36). These included cardiac, renal, limb, and facial anomalies, as well as genetic syndromes such as Beckwith-Wiedemann and pentalogy of Cantrell (37,38). Chromosomal anomalies are more commonly associated with omphalocele, especially small omphaloceles that do not contain liver (39,40). Babies with large or giant omphaloceles have an increased association with respiratory difficulties, including pulmonary hypoplasia and other pulmonary anomalies. The cause of the pulmonary hypoplasia is unknown. These infants have small narrow thoracic cavities and minimal pulmonary reserve. Mortality is high in these infants and reduction and repair in some babies may not be possible. Normal gastrointestinal (GI) motility and function is usual when the omphalocele sac is intact. Serositis is seen with ruptured omphaloceles. Lack of intestinal fixation is present in almost all these infants, and delayed complications may occur due to rotation.
In contrast to omphalocele, gastroschisis has a low incidence of associated anomalies. Intestinal atresia is seen in approximately 10% of infants with gastroschisis, and undescended testis may be present in up 30% of boys (41,42,43). It is not clear if this represents a common vascular etiology or the result of vascular compromise secondary to constriction of eviscerated bowel at the abdominal wall (44). Gastroschisis can result in severe abnormalities of fetal bowel function and prognosis of these infants is determined by the condition of the exteriorized bowel (7,45). Bowel damage results in morbidity through direct
interference with intestinal function and by leading to intrauterine growth retardation, amniotic fluid abnormalities, and preterm labor (46,47). The etiology of intestinal damage remains unclear. Animal models have been used to test several etiologic theories, including (1) damage is due to contact between the bowel and the amniotic fluid, and (2) damage is due to constriction of the bowel at the abdominal wall defect and may be partially dependent on exposure to meconium (48). In the chick embryo, bowel exposed to allantoic content but not amniotic fluid alone became thickened, edematous, and covered with a fibrous peel, suggesting that bowel damage is likely caused by urine in the amniotic fluid (49). This theory is supported by studies demonstrating that amnioallantoic fluid exchange can prevent damage in these models (50). Similarly, amniotic fluid-induced intestinal damage has been demonstrated in rabbit and sheep models. Mucosal absorption, decreased enzymatic function, altered gene expression, increased collagen production, and decreased smooth muscle function has been demonstrated in response to amniotic fluid exposure (51,52,53,54,55). Other studies have shown that the inflammatory peel is a result of amniotic fluid exposure and that bowel wall thickening and dilation is due to constriction at the abdominal wall defect. Moreover, the mechanism of constriction-induced bowel damage has been shown to result from mechanical obstruction and not ischemia (56,57). Bowel damage increases in severity with time of exposure to amniotic fluid, and both histologic and contractility changes are most severe toward the end of gestation (52,58).
interference with intestinal function and by leading to intrauterine growth retardation, amniotic fluid abnormalities, and preterm labor (46,47). The etiology of intestinal damage remains unclear. Animal models have been used to test several etiologic theories, including (1) damage is due to contact between the bowel and the amniotic fluid, and (2) damage is due to constriction of the bowel at the abdominal wall defect and may be partially dependent on exposure to meconium (48). In the chick embryo, bowel exposed to allantoic content but not amniotic fluid alone became thickened, edematous, and covered with a fibrous peel, suggesting that bowel damage is likely caused by urine in the amniotic fluid (49). This theory is supported by studies demonstrating that amnioallantoic fluid exchange can prevent damage in these models (50). Similarly, amniotic fluid-induced intestinal damage has been demonstrated in rabbit and sheep models. Mucosal absorption, decreased enzymatic function, altered gene expression, increased collagen production, and decreased smooth muscle function has been demonstrated in response to amniotic fluid exposure (51,52,53,54,55). Other studies have shown that the inflammatory peel is a result of amniotic fluid exposure and that bowel wall thickening and dilation is due to constriction at the abdominal wall defect. Moreover, the mechanism of constriction-induced bowel damage has been shown to result from mechanical obstruction and not ischemia (56,57). Bowel damage increases in severity with time of exposure to amniotic fluid, and both histologic and contractility changes are most severe toward the end of gestation (52,58).
TABLE 69-2 Abdominal Wall Defects. | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


