Wound Healing: Cellular and Molecular Mechanisms
Sundeep G. Keswani
Timothy M. Crombleholme
The Center for Molecular Fetal Therapy, The Division of General, Thoracic, and Fetal Surgery, Cincinnati Children’s Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, Ohio, 45229.
The Center for Molecular Fetal Therapy, The Division of General, Thoracic, and Fetal Surgery, Cincinnati Children’s Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, Ohio, 45229.
There is no physiologic function more essential to the surgeon’s craft than the innate ability of tissues to heal in response to injury, whether from trauma, burns, or a scalpel. For centuries, the management of wounds has been central to the function of the surgeon. In recent decades, there have been dramatic advances in our understanding of the cellular and molecular events that underlie the body’s response to injury and normal wound repair, as well as factors that are responsible for impairment of this process.
In this chapter, we first review the current understanding of the cellular and molecular events responsible for normal wound healing. We then examine the clinical application of wound healing in pediatric surgery, including the management of excessive scar formation. New insights into the biology of fetal wound healing has renewed interest in the quest for a regenerative response to injury, leading to the discovery of mechanisms of antiinflammatory cytokines and growth factors in the fetal environment. Last, we review two evolving areas of research in wound repair: the use of stem cells and gene transfer technology.
PHYSIOLOGY OF WOUND HEALING
Wound healing is the body’s attempt to restore normal structure and function to injured tissues. This occurs in some species, such as amphibians, through regeneration of injured or lost tissue. In postnatal humans, regeneration only occurs in the liver and the bone. In other tissues, repair is associated with varying degrees of scarring. Wound healing is a dynamic interactive sequence of growth factor and cytokine release, cellular migration and proliferation, matrix production and deposition, and remodeling (Fig. 12-1). These overlapping events characterize normal wound healing and by convention are temporally divided into four phases: hemostasis, inflammation, tissue proliferation, and tissue remodeling.
TISSUE INJURY
Although the mechanism of tissue injury may be considerably diverse, the sequence of the wound healing response is relatively constant in healthy patients. Injury disrupts the normal tissue architecture and, in higher vertebrate animals, the reparative response is fibroproliferative, rapidly creating a “patch” rather than regenerating the original tissue at the site of injury. Alterations in the normal reparative sequences may result in accentuated or protracted inflammation, which in turn leads to accentuation of tissue damage and repair. Dysfunctional wound healing is a characteristic of a range of conditions, such as diabetes, steroids, ischemia, venous hypertension, sepsis, and compromised nutrition, all of which may result in prolonged wound healing and can lead to chronic nonhealing wounds.
HEMOSTASIS
Formation of the Hemostatic Plug
Disruption of the normal tissue architecture with transection of blood vessels following injury, results in the exposure of blood elements to subendothelial collagen. This initiates coagulation by surface activation of Hageman factor, the release of tissue procoagulants from damaged cells and the exposure of phospholipids on activated platelets and endothelial cells (1). The key element that initiates the cascade is the surface absorption of coagulation proenzyme, which protects it from protease inhibitors and allows rapid amplification of the coagulation cascade leading
to clot formation. The extent of clot formation is also regulated at the microvascular level by several factors, including production of prostacyclin, which inhibits platelet aggregation (2); antithrombin III, which inhibits the activity of thrombin (3); protein C, which degrades coagulation factors V and VIII (4); and the release of plasminogen activator, which converts plasminogen to plasmin and initiates clot lysis (5). Plasminogen activator and plasmin are capable of degrading a large number of extracellular matrix proteins. Their activity is confined to cell surface microenvironment by the presence of plasminogen inhibitor bound to extracellular matrix protein (6).
to clot formation. The extent of clot formation is also regulated at the microvascular level by several factors, including production of prostacyclin, which inhibits platelet aggregation (2); antithrombin III, which inhibits the activity of thrombin (3); protein C, which degrades coagulation factors V and VIII (4); and the release of plasminogen activator, which converts plasminogen to plasmin and initiates clot lysis (5). Plasminogen activator and plasmin are capable of degrading a large number of extracellular matrix proteins. Their activity is confined to cell surface microenvironment by the presence of plasminogen inhibitor bound to extracellular matrix protein (6).
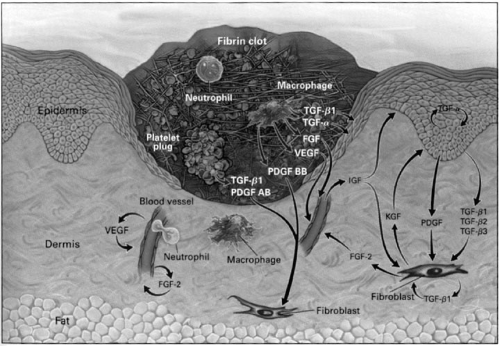 FIGURE 12-1. A cutaneous wound 3 days after injury. Wound healing is a dynamic interactive sequence of growth factor and cytokine release, cellular migration and proliferation, matrix production and deposition, and remodeling. (From Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999;341(10):738–746, with permission.) |
Once hemostasis has been established, a second major function of this primary hemostatic plug is to act as a provisional matrix, or a scaffold, for the recruitment of cells to the site of injury. Fibrin is a 340-kd hexamer composed of three fibrinogen chains that are coded on three different genes. Fibrinogen is synthesized in the liver and circulates in the blood. At the site of injury, thrombin cleaves the fibrinogen, which yields fibrin chains that polymerize with other fibrin molecules forming a fibrin polymer. The provisional network at this stage is unstable. Stability is achieved by the actions of plasma transglutaminases (factor XIIIa), which cause the formation of covalent intermolecular bonds by linking amino acid residues between adjacent fibrin chains resulting in a highly stable structure. This intricate network serves as a preliminary matrix that can bind and release cytokines and fibronectin. It also acts as a scaffold for migrating cells such as infiltrating monocytes, keratinocytes, endothelial cells, and fibroblasts (7). These migrating cells express integrins that bind to complimentary ligands on fibrin and fibronectin in the primary hemostatic plug (8). Fibronectin is composed of two 250-kd polypeptide chains linked by disulfide bonds. It expresses several domains that bind various macromolecules such as fibrin, collagen, heparin, and integrins expressed on migrating cells, which results in increased cell adhesion in the hemostatic plug.
Platelets
Platelets are among the first blood elements to arrive at the site of tissue injury. Platelet adhesion, activation, and degranulation can be stimulated by a number of factors, including adenosine diphosphate (ADP), thromboxane A2, epinephrine, platelet-activating factor (PAF), and exposure to subendothelial interstitial connective tissue. Platelets have specific cell surface receptors for each of the agonists and activation results in degranulation. Granules fuse with intracellular canaliculi to release their contents into the
extracellular environment in response to increased intracytoplasmic calcium level. The intracellular level of calcium is cleared by cyclic adenosine monophosphate (cAMP) via phosphorylation of an adenosine triphosphate (ATP)-dependent calcium pumping system and therefore inhibits platelet function (9,10). Activation results in discharge of the alpha granules that release fibrinogen, fibronectin, thrombospondin, and von Willebrand factor VIII among other factors. Von Willebrand factor VIII mediates platelet adhesion to fibrillar collagen while the other three initiate platelet aggregation. Activation also results in the expression of various glycoproteins such as the GPIIb/IIIa surface receptor, which mediates the above four adhesive proteins and interactions between platelets and endothelium that results in platelet aggregation and adhesion (11). Platelets also have other types of granules that contain molecules that participate in wound repair. Platelet dense bodies contain serotonin, calcium, ATP, ADP and pyrophosphate. Platelets produce thromboxane A2, a derivative of arachiodonic acid that has potent vasoconstrictor and platelet aggregative abilities. In addition, alpha granules discharge growth factors into the primary hemostatic plug such as platelet-derived growth factor (PDGF), transforming growth factor alpha (TGF-α), and transforming growth factor beta (TGF-β), which each contribute to cellular recruitment and migration, granulation tissue formation, matrix deposition, and directly or indirectly to neovascularization and reepithelialization.
extracellular environment in response to increased intracytoplasmic calcium level. The intracellular level of calcium is cleared by cyclic adenosine monophosphate (cAMP) via phosphorylation of an adenosine triphosphate (ATP)-dependent calcium pumping system and therefore inhibits platelet function (9,10). Activation results in discharge of the alpha granules that release fibrinogen, fibronectin, thrombospondin, and von Willebrand factor VIII among other factors. Von Willebrand factor VIII mediates platelet adhesion to fibrillar collagen while the other three initiate platelet aggregation. Activation also results in the expression of various glycoproteins such as the GPIIb/IIIa surface receptor, which mediates the above four adhesive proteins and interactions between platelets and endothelium that results in platelet aggregation and adhesion (11). Platelets also have other types of granules that contain molecules that participate in wound repair. Platelet dense bodies contain serotonin, calcium, ATP, ADP and pyrophosphate. Platelets produce thromboxane A2, a derivative of arachiodonic acid that has potent vasoconstrictor and platelet aggregative abilities. In addition, alpha granules discharge growth factors into the primary hemostatic plug such as platelet-derived growth factor (PDGF), transforming growth factor alpha (TGF-α), and transforming growth factor beta (TGF-β), which each contribute to cellular recruitment and migration, granulation tissue formation, matrix deposition, and directly or indirectly to neovascularization and reepithelialization.
Vascular Response to Injury
The initial vascular response to injury is vasoconstriction, which serves to control hemorrhage. This is initially mediated by action potentials initiated by injured blood vessels and then maintained by the release of epinephrine, serotonin, and thromboxane A2 by platelets. Once the initial danger of exsanguination is controlled, the vasculature vasodilates to allow the mass influx of cells required for the repair of tissue. This is mediated by histamine derived from mast cells (12). Further vasodilation is caused by the release of the cyclooxygenase products, prostaglandin E1 and E2, bradykinin, and endothelial-derived nitric oxide. The vascular response is also mediated by factors released by the primary hemostatic plug, such as Hageman factor (13), which initiates the classical and alternative complement cascades. Generation of C3a and C5a enhance vascular permeability, and also serve as chemoattractants for neutrophils and monocytes (14).
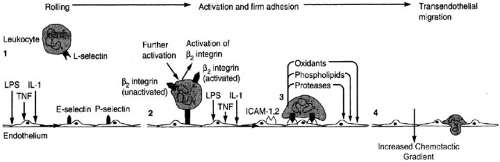 FIGURE 12-2. The process of leukocyte recruitment into areas of inflammation. At first, the cell exhibits a rolling type of motion due to loose adhesion between L selectin and E selectin. Progressive activation results in the expression of more adhesion molecules, firmer connections between the leukocyte and the endothelial surface, and finally transendothelial leukocyte migration. (From Lee G. Wintrobe’s clinical hematology, 10th ed. Philadelphia: Lippincott Williams & Wilkins, with permission.) |
INFLAMMATION
During the inflammatory phase devitalized tissues are debrided, and the milieu is conditioned for the enormous mobilization of cellular and noncellular elements to the repair site. The successful completion of this phase is vital to the subsequent repair process. The regulation of this phase is equally important because an uncontrolled inflammatory response can rapidly spread beyond the boundaries of a local reaction and engulf the affected organism in a life-threatening inappropriate systemic response.
Drawn by the release of chemoattractants from aggregating platelets and wounded tissue, numerous inflammatory cells infiltrate the wound site. Each specific type of infiltrating inflammatory cell appears with a predictable time course. Early in the inflammatory period, polymorphonuclear leukocytes are the predominant infiltrating cells. Leukocytes migrate to the site of injury from the circulatory system by diapedesis, which is initiated by adhesion molecules expressed on the activated endothelium and leukocytes (Fig. 12-2). Initial contact is between the E selectin on the endothelial cell and the L selectin on the leukocyte, which act to slow down the movement of the leukocyte and induce a rolling pattern to bring it closer to the periphery of the lumen of the blood vessel. It is the
interaction between the integrins of the leukocyte and the intercellular adhesion molecule of the endothelial cell that halts the progression of the leukocyte and allows the diapedesis of leukocytes between endothelial loose junctions to migrate to the site of injury along a chemotactic gradient (15).
interaction between the integrins of the leukocyte and the intercellular adhesion molecule of the endothelial cell that halts the progression of the leukocyte and allows the diapedesis of leukocytes between endothelial loose junctions to migrate to the site of injury along a chemotactic gradient (15).
Neutrophils
Neutrophils flood the wound early in the healing response and are recruited to the wound by many factors, including cleavage products of fibrinogen, thrombin, fibrin split products, plasmin, C5a, leukotriene B4, PDGF, PAF derived from endothelial cells, and N-formyl peptides released from bacteria (16). The neutrophil is a myeloid lineage cell that is derived from the bone marrow that can persist in the wound for 1 to 2 days, does not replicate, and is rapidly cleared. An increase in neutrophil expression of CD11/CD18 mediates adherence of neutrophils to blood vessel endothelium, facilitating extravasation of neutrophils at the site of injury (17). The primary role of neutrophils in wound healing is phagocytosis and destruction of bacteria via oxygen-free radicals and enzymatic mechanisms. The nicotinomide adenine dinucleotide phosphate system produces the toxic oxygen radicals that facilitate elimination of ingested microbes (18). Most neutrophils become trapped in the desiccated tissue or wound clot and slough off during the tissue repair process. The remainder become senescent and are phagocytosed by tissue macrophages.
Monocytes
As neutrophils cleanse the wound, they are either sloughed off in the overlying eschar or become senescent within the wound bed. Monocytes are recruited by monocyte specific chemoattractants, including collagen fragments, elastin, fibronectin, thrombin, and TGF-β and chemokines. Monocytes continue to accumulate, reaching a peak at approximately 3 days postinjury, and replace neutrophils as the dominant inflammatory cell found in the wound. Once the tissue monocyte has left the circulatory system, they are known as macrophages. Macrophages bind to extracellular matrix proteins through integrin receptors, stimulating extracellular matrix phagocytosis and digestion of tissue debris, bacteria, and senescent neutrophils. To enhance debridement, macrophages release enzymes such as collagenase.
The adherence of monocytes to extracellular matrix proteins also alters gene expression, converting them to activated macrophages with the induction of colony-stimulating factor-1 and tumor necrosis factor-alpha (TNF-α). TNF-α acts locally to stimulate endothelium and activate thromboplastin (19). In addition, macrophages become an abundant source of growth factors, such as PDGF, TGF-β, TGF-α, insulin like growth factor 1 (IGF-1), interleukin 1 (IL-1), and fibroblast growth factor (FGF), which are essential in the propagation of new matrix deposition, angiogenesis, stem cell recruitment, and reepithelialization. The tissue macrophage is believed to play a pivotal and essential role in the transition from inflammation to repair. The ultimate goal of these actions is recruitment of fibroblasts, which are necessary for the deposition of collagen and restoration of tensile strength in the injured tissue.
Angiogenesis
Essential to the maintenance of the repair process and the resulting newly synthesized tissue is a delivery mechanism for nutrients. Early on, diffusion and cellular migration supply the wound site; however, new vasculature must develop to maintain the delivery of cells active in repair, provide them with adequate nourishment, and remove the by-products of metabolism. This need is met by the formation of new blood vessels.
TISSUE PROLIFERATION
The onset of angiogenesis provides the injured wound a means to meet its metabolic requirements and a steady influx of cells. The wound has been cleaned and is now ready for the next stage in tissue repair. Fibroblasts, migrating in response to chemoattractants such as TGF-β and PDGF-B, populate the wound site in increasing numbers throughout the first two phases of wound healing. Initial activities center around the formation of granulation tissue and deposition of extracellular matrix (ECM) around which the reparative architecture is constructed. Later in the repair process, the fibroblast is responsible for the deposition of new collagen. Along with this accumulation of collagen-producing cells, this phase is characterized by the reepithelialization of the wound.
Granulation Tissue
The angiogenic process is accompanied by the formation of granulation tissue, which in addition to the new blood vessels is composed of macrophages, fibroblasts, and loose connective tissue. Endothelial cells, macrophages, and fibroblasts have highly orchestrated roles in the formation of new granulation tissue. Endothelial cells form capillaries that provide a source of oxygen, nutrients, and cells.
Tissue macrophages secrete growth factors and cytokines that attract fibroblasts, and fibroblasts produce the extracellular matrix. In addition, the hemostatic plug also promotes the formation of granulation tissue by acting as a scaffold for the provisional matrix formation, being a reservoir for cytokines and through a direct action of matrix-induced integrin cell signaling, which facilitates fibroblast migration (23).
Tissue macrophages secrete growth factors and cytokines that attract fibroblasts, and fibroblasts produce the extracellular matrix. In addition, the hemostatic plug also promotes the formation of granulation tissue by acting as a scaffold for the provisional matrix formation, being a reservoir for cytokines and through a direct action of matrix-induced integrin cell signaling, which facilitates fibroblast migration (23).
Fibroblasts migrate into the wound in response to growth factors such as PDGF-B and FGF, which also induce fibroblast proliferation and the production of ECM. There is a mutual interaction between fibroblasts and the ECM. Fibroblasts move through the ECM by integrin receptors, and continue to produce and remodel ECM proteins through the secretion of proteolytic enzymes, such as the matrix metalloproteinases (24). In turn, the ECM regulates, in part, the production of new ECM by the fibroblast (25). This relationship between ECM and fibroblasts is integral to the formation of new granulation tissue.
Once the fibroblasts have migrated into the wound, their major role turns to protein synthesis. This results in the high-yield synthesis and deposition of collagen, as well as the production of growth factors. Once an abundant collagen matrix has been deposited, collagen production by fibroblasts is downregulated (26). Approximately 10 days postinjury, fibroblasts begin to undergo apoptosis and mark the beginning of the transition to the hypocellular scar.
Extracellular Matrix Proteins
The ECM may prove to be a critical factor in determining the type of repair that occurs in wounded tissue. This complex framework consists of glycoproteins, glycosaminoglycans, and proteoglycans, which are relatively complex macromolecules composed of varying proportions of modified peptide and carbohydrate moieties. Proteoglycans consist of repeating polysaccharides around a protein core. Glycosaminoglycans make up the polysaccharide chain in the proteoglycan. These macromolecules can avidly bind water and cations, forming a gellike substance. Functions of the proteoglycans in the wound include regulation of blood coagulation, extracellular matrix organization, and acting as a reservoir of growth factors and cytokines.
The first role of the ECM in tissue repair is blood coagulation. Heparin and heparan sulfate are released from mast cells and act by potentiating the anticoagulation effects of antithrombin III and thrombin (27). Subsequently, these mast cells release histamine, which results in vasodilation to facilitate the influx of cells and nutrients. These macromolecules also serve to give organization to the newly forming wound bed and then provide direction for the influx of infiltrating cells. Proteoglycans containing chondroitin 4 sulfate and dermatan sulfate are produced by fibroblasts and serve as structural components of the evolving scar (28). Dermatan sulfate is increased in granulation tissue and collagen deposition. It has also been suggested that proteoglycans regulate collagen fibrillogenesis (29). Both collagen synthesis and chondroitin 4 sulfate are elevated in hypertrophic scars that support this hypothesis. Hyaluronic acid, a glycosaminoglycan, has a large water shell that enhances cell migration and in concert with fibrin acts to provide a scaffold for migrating fibroblasts in the early wound matrix (30). Glycoproteins such as fibronectin act as adhesive molecules or the “glue” in the wound that assists in cell attachment and migration. The major extracellular matrix proteins and their actions are summarized in Table 12-1.
Collagen Structure and Deposition
All wound extracellular matrices contain collagen as their primary component. The biology and chemistry of this ubiquitous compound have been well characterized. At least 25 types of collagen have been described; characteristics of the more common types are listed in Table 12-2.
The collagens have as their common structure a triple helical conformation composed of similar but nonidentical polypeptides. A high frequency of the tripeptide sequence glycine–proline–x is demonstrated, with x being hydroxyproline in about 10% of the sequences. The heavy glycine content is essential to the helical structure that predominates in the collagens. Glycine, the smallest amino acid, with a side chain composed of a single hydrogen atom, provides a chemical swivel around which the repeated coiling can occur (31).
The types of collagen in tissues have been closely correlated with the function of these tissues. Type I collagen imparts a rigid structure to tissues and is therefore the predominant collagen found in bone. Type III collagen is found in elastic structures, constituting as much as 70% of the total collagen content in large arteries. Skin exhibits properties of both strength and elasticity, and its collagen makeup is about 80% type I and 20% type III. Of the remaining collagens, type II is the predominant collagen in cartilage, whereas type IV is found primarily as a component of basement membranes.
Collagen is assembled in a complex series of steps that require numerous cofactors for proper synthesis. Before collagen is secreted from the cell, hydroxylation of proline and lysine residues by prolyl and lysyl hydroxylase occurs. Oxygen, ferrous iron, α-ketoglutarate, and ascorbic acid are cofactors critical to the proper function of the hydroxylases catalyzing these events. In addition, manganese is required for the subsequent glycosylation of hydroxylysine residues. The essential nature of these cofactors is highlighted by the effects of deficiencies of these cofactors, which can result in impaired wound healing secondary to the rapid breakdown of unstable collagen structures in the ECM.
TABLE 12-1 The Glycosaminoglycans. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 12-2 Major Types of Collagen. | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
After the intracellular formation of collagen in fibroblasts and the subsequent hydroxylations of the amino acid residues, the collagen is secreted into the ECM in its triple helical form as procollagen, with a number of peptides, termed propeptides, remaining at both the carboxy and amino terminals. Orientation of the procollagen molecules are maintained by orientation of fibroblasts along the direction of fibrin-fibronectin strands known as traction morphogensis. The propeptides are then rapidly cleaved from procollagen, resulting in tropocollagen. At this point, lysyl oxidase, a copper-containing enzyme, catalyzes the formation of stable intermolecular and intramolecular cross-links. This results in a longitudinal alignment of the collagen molecules into fibrils, yielding a characteristic overlap pattern with a periodicity of 68 nm when viewed by electron microscopy. These cross-links are critical to normal collagen synthesis. Deficient collagens may result when pharmacologic agents such as β-amino propionitrile or D-penicillamine are administered. β-Amino propionitrile impairs collagen synthesis directly through its inhibition of lysyl oxidase, and D-penicillamine impairs collagen synthesis by binding to collagen at the sites of cross-link formation, thereby inhibiting their development (32,33).
Reepithelialization
Although the neutrophil provides temporary defense against invasion through the breach of the wounded integument, surface closure of the defect also must be achieved. Within hours of injury epithelial cells have begun to move across the wound. In the skin, keratinocytes do not appear to move along single file but appear to “leapfrog” over each other while advancing across the wound (34). During this migration, epithelial cells undergo a morphologic change that results in a loosening of attachments from the surrounding cells and basement membrane, which allows for greater lateral mobility and an increased ability to reepithelialize the wound. The migrating epidermal cells have a unique phenotype that resembles growing keratinocytes in culture. The induction signals for these migrating cells are unknown. It has been hypothesized that in the free edge of a wound, a loss of contact inhibition and mitogenic and motogenic growth factors may each play a role as key induction signals. Possible growth factors for the induction of keratinocytes include are Keratinocyte growth factor (KGF), Epidermal growth factor (EGF), and TGF-α (35). Research in transgenic animals in which these growth factors are knocked out or overexpressed demonstrate marked changes in reepithelialization, which suggests these growth factor have a significant role in wound reepithelialization (36).
If the basement membrane is injured, wound keratinocytes migrate over a provisional matrix composed of type V collagen, fibrin, fibronectin, vitronectin, and tenascin. The fibrin and fibronectin is initially derived from the circulation, but is later supplied by cells of the provisional matrix, including fibroblasts and macrophages. Frequently, this process is not on top of the wound, but between the wound surface and the newly developed eschar consisting of desiccated cells and nonviable tissue. As the migrating cells course this plane, they release collagenases that permit migration over the wound.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


