Undue Susceptibility to Infection
Talal A. Chatila
Each year, a large number of children are evaluated by their primary care physicians for recurrent infections, an especially common event in early childhood.1 The overwhelming majority of such cases are benign, and an extrinsic cause of recurrent infection is identified. Examples of extrinsic causes include heightened exposure to pathogens in a daycare setting, carriage of a pathogenic organism such as Staphylococcus aureus in the context of recurrent infection with this organism, or recurrent upper respiratory tract infections in the context of parental smoking. However, concern about an intrinsic pathologic underpinning is heightened on the basis of frequency of infections, their severity, and the nature of the offending organism. The coexistence of multisystem disease, autoimmunity, or lymphoreticular malignancy should also prompt evaluation for immunodeficiency. A family history of recurrent infections raises the index of suspicion.
One helpful clue to the presence of a host abnormality is a high frequency of infections. Examples include two or more systemic bacterial infections at any time (such as sepsis, deep-seated abscesses, or meningitis), three or more bacterial infections (eg, draining otitis media), or six to eight or more upper respiratory tract infections in 1 year.2 The last finding should be modified by the fact that many children, especially toddlers, suffer from recurrent upper respiratory tract infection from repeat exposure to respiratory (usually viral) pathogens, especially during the first year of daycare attendance.
Recurrent infections with a particular organism also point to abnormalities of the host. A case in point is meningococcemia, in which a second episode of this disease raises the prevalence of a terminal complement pathway abnormality in afflicted individuals from ≤1% to 30 to 40%. Other examples include Staphylococcus aureus infections in children with chronic granulomatous disease (CGD) or leukocyte adhesion deficiency (LAD).
The severity of the recurrent infection is reflective of the seriousness of the underlying disorder. The compromised child may fail to recover completely between infections. Failure to thrive, weight loss, and growth retardation are grave manifestations of immunodeficiency and call for immediate investigation. The need for surgical intervention provides yet another measure of the severity of the underlying infection. Such interventions may include myringotomy tube placement for chronic otitis media, sinus surgery for chronic sinusitis, lobectomy for chronic right middle lobe pneumonia, and drainage of superficial and deep abscesses.
The availability of effective antibiotic therapy may modulate the presentation, but in general the clinical picture may slowly but progressively worsen over a protracted period of time.
The nature of sites affected by the recurrent infections also provides valuable clues to the problem at hand. Humoral immunodeficiency, cystic fibrosis, and immotile cilia syndrome result in recurrent severe sinopulmonary infections including chronic sinusitis, pneumonia, and bronchiectasis. Humoral immunodeficiency may also result in chronic diarrhea as a consequence of infestations with pathogens such as Giardia lamblia. Recurrent infections affecting one particular site (eg, one specific lung lobe or one ear) may point to underlying anatomic abnormalities.
Recurrent upper respiratory infections in the absence of other organ system involvement may point to allergic disorders or to mild atypical forms of cystic fibrosis. Supporting evidence may come from a family history of allergic diseases, including asthma and atopic dermatitis, and findings on physical exam of allergic attributes such as allergic shiners, clear nasal discharge eczema, or wheezing. Diagnosis can be established by appropriate testing including allergen skin testing, serum IgE, and sweat test for cystic fibrosis. A single infection with Pneumocystis jiroveci is sufficient grounds for a careful search for an immunodeficiency. Patients with asplenia, whether acquired in the course of sickle-cell disease or more rarely congenital, may present with sepsis with encapsulated organisms.
Restricted sets of pyogenic pathogens also characterize other diseases. Patients with cystic fibrosis are particularly susceptible to respiratory infections with Pseudomonas aeruginosa, Burkholderia sp., and Staphylococcus aureus. Those with chronic granulomatous disease suffer from recurrent infections with catalase-positive organisms including Staphylococcus aureus, Chromobacterium violaceum, Pseudomonas cepacia, Serratia marcescens, Candida, and Aspergillus species.
Defects in cellular immunity predispose to recurrent, often disseminated viral infections, especially with herpes viruses. Patients with cellular immune defects may also suffer from atypical mycobacterial infections, mucocutaneous candidiasis, and Pneumocystis carinii pneumonia.
UNDUE SUSCEPTIBILITY TO SPECIFIC PATHOGENS
Defects in the adaptive and innate immune response may also give rise to susceptibilities to specific infection (see Tables 187-1 and 229-1).3 Hypomorphic (partially penetrating) mutations in the CD40 ligand (CD40L) result in undue susceptibility to chronic parvovirus infection, whereas the more severe mutations in CD40L or its counter-receptor CD40 give rise to opportunistic infections with P jiroveci, Cryptosporidium, and Toxoplasma. Mutations in the signaling lymphocyte activation molecule-associated protein and the X-linked inhibitor of apoptosis give rise to the clinically related syndromes of X-linked lymphoproliferative disease, with peculiar susceptibility to Epstein-Barr virus infection.
Narrowly focused susceptibility to infections is more likely to result from defects in the innate immune response (Table 187-1).3 A predilection toward infections with neisserial species with deficiency of terminal complement components and, to a lesser extent, in components of the alternate pathway of complement activation. Phagocytic cell defects typically present with recurrent pyogenic and fungal infections. A particular form of phagocytic cell defect, chronic granulomatous disease, predisposes the individual to infections with catalase-positive organisms. Mutations in the IL-12/IL-12/IL-23 receptor interferon gamma receptor/STAT1 axis give rise to undue susceptibility to mycobacterial infections. Deficiency of the adaptor protein MYD88 and the downstream kinase interleukin-1 receptor-associated kinase 4, both coupled to several Toll-like receptors, results in invasive infections with pyogenic and encapsulated organisms. Deficiency of the MYD88-independent Toll-like receptor (TLR) 3 and of the endoplasmic reticulum resident membrane protein UNC93, which is involved in intracellular trafficking of TLR3 and the nucleotide-sensing receptors TLR7-9, results in peculiar susceptibility to herpes simplex encephalitis, which is caused by an infection with an individual pathogen (herpes simplex).4,5 Although this infection may occur only once in a lifetime and was previously thought to be due to a random event, it is now understood that this infection represents a specific host susceptibility. Undue susceptibility to papilloma viruses have been uncovered in two syndromes: WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis), resulting from mutations in the chemokine receptor CXCR4 syndrome, and epidermodysplasia verruciformis, resulting from mutations in the keratinocyte proteins EVER1/2.
Table 187-1. Genetic Defects Associated with Undue Susceptibility to Specific Pathogens
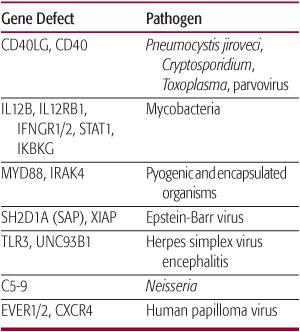
Gene symbol and name: CD40LG, CD40 ligand; IL12B, interleukin 12 p40 subunit; IL12RB1, IL12 receptor b chain; IFNGR1/2, interferon γ receptor subunit 1/2; STAT1, signal transducer and activator of transcription 1; IKBKG: inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma; MYD88: myeloid differentiation factor 88; IRAK4, interleukin-1 receptor associated kinase 4; SH2D1A (SAP), SLAM-associated protein; XIAP, X-linked inhibitor of apoptosis; UNC93B1, Unc-93 homolog B1; TLR3, Toll-like receptor 3; EVER1/2, Epidermodysplasia verruciformis 1, 2 genes; CXCR4, chemokine (C-X-C motif) receptor 4.
INVESTIGATION OF THE CHILD WITH UNDUE SUSCEPTIBILITY TO INFECTIONS
The evaluation for immunodeficiency is directed by the clinical presentation, including consideration of the specific type of pathogens involved and the resultant suspected immunodeficiencies.6,7 Secondary immunodeficiencies such as HIV infection can present in a fashion similar to primary immunodeficiency disorders and should be ruled out.
A family history of severe or fatal infections in a maternal uncle suggests an X-linked immunodeficiency disorder such as chronic granulomatous disease or severe combined immunodeficiency (SCID) in boys, whereas easy bruising in a boy with recurrent infection is suggestive of another X-linked disorder, the Wiskott-Aldrich syndrome.
The physical exam may provide clues that raise concerns, including findings of failure to thrive, the presence of allergic stigmata including allergic shiners, eczema, wheezing, or the presence of thrush. Absent tonsillar tissue (barring tonsillectomy) points to failure of B-cell development, which can be confirmed by the absence of an adenoid shadow on lateral neck view radiographic images. Characteristic facial features and cardiac murmurs may result from DiGeorge syndrome, and dwarfism may point to immunodeficiency associated with the cartilage-hair hypoplasia syndrome.
Documentation of infectious episodes by testing for pathogens (cultures, serologies, or DNA testing) or radiographically (sinus/chest x-rays, etc) is most useful in confirming the problem of recurrent infections and provides clues to pathogenesis (including anatomical abnormalities) and therapy.
Investigations useful in the child with undue susceptibility to infection are shown in Table 187-2. A complete blood count with differential is useful to rule out immunodeficiencies associated with lymphopenia or neutropenia. A sweat test screens for cystic fibrosis, and allergen skin testing helps discern atopy-associated recurrent upper airway infections that may result in recurrent infection from causes other than immunodeficiency. An elevated erythrocyte sedimentation rate would be consistent with a history of recurrent infections. Quantitation of total immunoglobulin levels is useful to rule out hypogammaglobulinemia.
Functional antibody responses to protein and polysaccharide antigens can be assessed in the immunized individual by measuring specific antibody titers to tetanus toxoid and to pneumococcal serotypes represented in the polyvalent pneumococcal vaccine, respectively. Response to the polyvalent pneumococcal vaccine may not be assessed before age 2 because the vaccine elicits poor response in young infants and children. The integrity of the classic pathway of complement activation can be assessed by measuring the total hemolytic activity in the blood (CH50) (see Chapter 189). Cellular immunity can be assessed by delayed-type hyper-sensitivity skin mitogen stimulation assays and by flow cytometric analysis of T-lymphocyte markers.
Additional tests may be performed when suspicion of an underlying abnormality is high. Recurrent infections with catalase-positive organisms suggests the diagnosis of chronic granulomatous disease. Persistent thrush or the isolation of organisms such as cytomegalovirus and P jiroveci would point to defective cellular immunity. The total hemolytic complement activity (CH50), as well as serum levels of specific complement components, can be measured if an association with autoimmunity is suspected. Recurrent neisserial infections may point to defects in the terminal complement components (screened by the CH50 test) or in the alternate pathway of complement (screened by a modified CH50 assay and by measuring levels of alternate pathway component factors B and D). A high index of suspicion for cystic fibrosis in the face of normal sweat test results may prompt DNA genotyping for unusual mutations in the cystic fibrosis transporter gene, whereas unrelenting sinus and lung infections may prompt performance of a biopsy for ciliary abnormalities.
Table 187-2. Investigation of the Child with Undue Susceptibility to Infections



