Disorders of Metal Metabolism
Jean-Marie Saudubray
Metals are indispensable elements of cell biology. They function as cofactors in many specific proteins and are involved in all major metabolic pathways. Their metabolism and implications in inborn errors of metabolism are still not well known, but the number of inherited metabolic disorders involving the absorption, transport, or metabolism of metals is rapidly growing. Clinical presentations are very diverse and can involve all organs and systems, including the liver and the central nervous system (Table 169.1). Deficiency in metals results in metabolic abnormalities due mostly to loss of function of metal-dependent proteins. On the other hand, excess of metals can result in the unregulated oxidation of proteins, lipids, and other cellular components, causing subsequent tissue injury. Some inherited metal disorders are treatable by chelating drugs or by daily supplementation of the missing metal at pharmacological doses.
COPPER
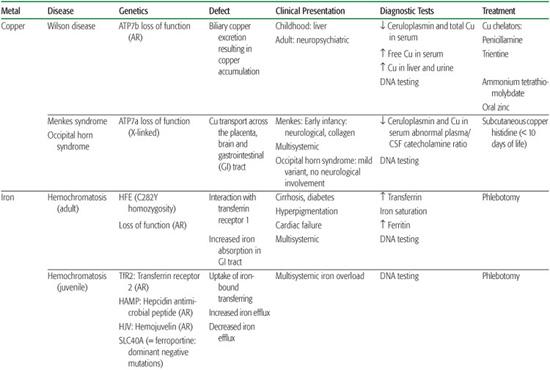
Copper is readily available in the diet. After absorption through the stomach and duodenum, it is rapidly removed from the portal circulation by hepatocytes. Biliary excretion is the only physiological mechanism of copper elimination, and at steady state, the amount of copper excreted into the bile is equivalent to that absorbed from the intestine.1 Ctr1 is a plasma membrane protein essential for early embryonic development, intestinal copper uptake, and transportation from the plasma into the brain.2 Ninety percent of plasma copper is bound to ceruloplasmin. Intracellular copper metabolism is dependent on the copper transport ATPases, ATP7a, and ATP7b. Intracellular trafficking also requires proteins called metallochaperones (superoxide dismutase, Atox1, CCS, Cox 17, Sco1, and Sco2), which direct copper to specific cellular pathways.3 Wilson disease or hepatolenticular degeneration is due to mutations in the ATP7B gene, which encodes a copper-transport protein required for exporting copper from the liver into the bile. Menkes disease is due to mutations in the ATP7A gene, which encodes a copper-transport protein required for the efflux of copper from cells (see Chapter 573).
IRON
Ferrous ion (Fe2+) is taken up into cells by the divalent membrane transporter DMT1, whereas ferric iron (Fe3+) enters cells (including brain cells) via endocytosis of the transferrin receptor following binding to transferrin.4 Iron requirements far exceed the gastrointestinal absorption capacity; thus, almost all the iron utilized each day is continuously recycled from internal stores.5 Steap3 is a ferrireductase critical for transferring-mediated iron release into the cell. Ferritin is the predominant storage protein consisting of heavy chains with ferroxidase activity and light chains. Ferroportin is the only cellular iron exporter, and ceruloplasmin is a ferroxidase that mediates efficient cellular iron release. Iron homeostasis is regulated by hepcidin, a circulating peptide that binds to ferroportin, mediating uptake and degradation of this exporter.
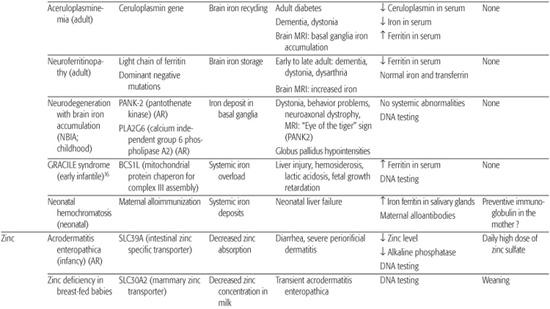
Iron enters mitochondria via mitoferrin, and frataxin is a mitochondrial protein mediating Fe-S cluster formation and heme biosynthesis. Although many of the proteins essential for systemic iron homeostasis are expressed within the brain, genetic disorders resulting in loss of functions of these proteins rarely result in brain iron overload or deficiency or neurological disease. Conversely, the concentration of iron in the brain is greatest in the basal ganglia and in astrocytes; this supports the idea that these cells function in iron storage and regulation. Aceruloplasminemia, neuroferritinopathies due to ferritin light chain gene mutations, and neurodegeneration with brain iron accumulation are recent examples of this brain iron metabolism defect6 (Table 169.1; see Chapter 573).
ZINC
Zinc is a cofactor for over 100 enzymes and as such is involved in all major metabolic pathways. It is also essential for nucleic acid metabolism and protein synthesis and their regulation through zinc-finger proteins. In humans, about 1% of the total body zinc content is replenished daily by the diet.7 This is accomplished principally by tight control of two systems: absorption from the intestine and endogenous loss via pancreatic and other intestinal secretions. The zinc transporter Znt proteins lower intracellular zinc by mediating zinc efflux from cells or influx into intracellular vesicles. SLC30A mutations are responsible for zinc deficiency in breast-fed babies.8 The Zip proteins promote zinc transport from the extracellular fluid or from intracellular vesicles into the cytoplasm. SLC39A mutations are responsible for acrodermatitis enteropathica.9 Zinc deficiency, either hereditary or acquired, has major detrimental effects, whereas high serum zinc has few, probably because of binding to albumin or a2-macroglobulin.
Table 169-1. Inborn Errors of Metal Transport and Metabolism
MAGNESIUM
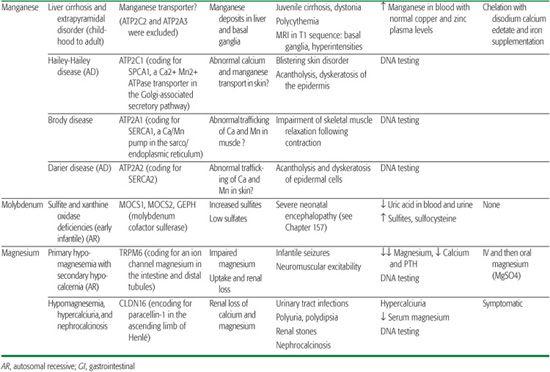
Magnesium is the second most important intracellular cation and plays an essential role in many biochemical processes and in neuromuscular excitability. Its homeostasis is regulated by the interplay between intestinal absorption and renal excretion.
MANGANESE
Physiology of manganese homeostasis remains poorly understood, but it seems certain that concentrations in subcellular compartments are closely controlled despite quite large variations in dietary manganese intake (eg, it is higher in vegetarians). Manganese plays an important role in many biological processes. It acts as a cofactor for various enzymes in the cytoplasm, Golgi apparatus, and mitochondria. However, high cytoplasmic concentrations of manganese are cytotoxic. Two Ca2+-ATPases are known to be involved in Mn2+ transport: the Golgi-associated secretory pathway (SPCA) and the sarco/endoplasmic reticulum (SERCA)Ca2+ ATPases.10,11 They transport both calcium and manganese. Mutations in ATP2C1, a gene encoding for SPCA1, are responsible for Hailey-Hailey disease11 (OMIM 169600). ATP2C2, which encodes for SPCA2 (a protein thought to be the main candidate for manganese uptake and detoxification via the secretory pathway), was excluded in the cirrhosis/dystonia syndrome.12 Three human SERCA pumps—SERCA1, SERCA2, and SERCA3—encoded by ATP2A1, ATP2A2, and ATP2A3 genes are known. Mutations in the ATP2A1 gene are responsible for Brody disease13 (OMIM 601003), and mutations in the ATP2A2 gene are known to cause Darier disease14 (OMIM 124200). Several of the proteins involved in manganese uptake and transport are shared with those involved in iron intake and transport.15 It is tempting to speculate that similar mechanisms of iron regulation by hepcidin could also exist for manganese homeostasis.12
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


