Physiologic changes of the hemostatic system in pregnancy
Understanding disease in pregnancy requires knowledge of the normal physiologic changes occurring in pregnancy (Table 4.1). The hemostatic system is a physiologic system designed to limit bleeding at sites of vascular injury. Hemostasis, involving vasoconstriction, platelet plug formation through platelet aggregation, fibrin clot formation through coagulation activation, and fibrinolysis or clot dissolution, is normally limited to sites of vascular injury. Each of these processes is classically thought to occur in sequence (in phases) but a more modern understanding is that these steps occur in varying sequence and at varying rates in different vascular beds in a time, flow and interdependent manner. The hemostatic system undergoes many physiologic changes in an effort to prepare the parturient for the hemostatic challenge of delivery. These changes have likely evolved due to the tremendous selective pressure from maternal mortality caused by hemorrhage at childbirth throughout evolution. As recently as the 17th century, more than 10% of parturients died at childbirth from hemorrhage.
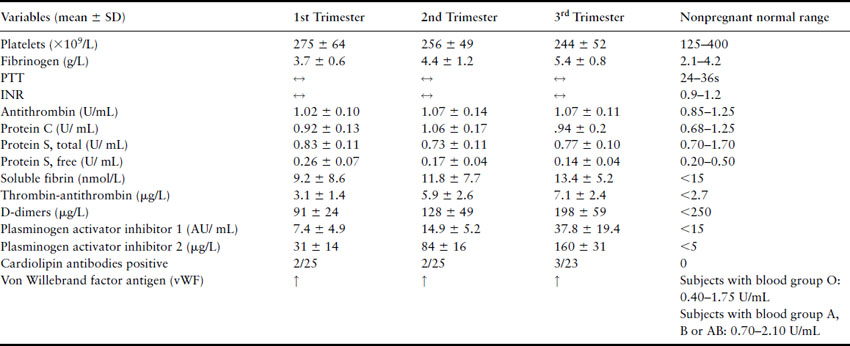
Table 4.1 Hemostatic changes in pregnancy. Adapted from Bremme [1]
Hypercoagulability of pregnancy
Pregnancy is associated with increased levels of procoagulant factors and decreases in anticoagulant factors, resulting in increased thrombin generation [2,3]. This increased thrombin generation coupled with a decrease in fibrinolysis results in hypercoagulability that prepares expectant mothers for the hemostatic challenge of delivery. Progressively throughout gestation, some procoagulant factors including factor VIII, factor V and fibrinogen increase while the anticoagulant mechanisms are reduced, including increasing resistance to activated protein C and reduced protein S levels. Antithrombin and protein C levels are not reduced in pregnancy [2]. The gradual increase in thrombin activation throughout gestation is evidenced by increases in coagulation activation markers including thrombin antithrombin complex (TAT) levels and F1.2 (a byproduct of thrombin acting on fibrinogen) [4]. Von Willebrand factor (vWF) is involved in two components of the hemostatic mechanism: coagulation, in which vWF acts as a plasma carrier protein for FVIII, and platelet plug formation where vWF permits adhesion of platelets to the subendothelial matrix proteins. Von Willebrand factor levels also increase throughout pregnancy. Fibrinolytic activity (mediated by plasmin) is reduced in pregnancy most likely due to increased levels of plasminogen activator inhibitor (PAI) type 1 and type 2 (produced mainly by the placenta). Thrombin activatable fibrinolysis inhibitor (TAFI) has been demonstrated to be a key component of the reduced fibrinolysis seen with increasing gestation [5]. D-dimers are byproducts of fibrinolysis created by the action of plasmin on cross-linked fibrin. These increasing D-dimer levels may not appear consistent with the previously mentioned decreased fibrinolysis observed in pregnancy but are likely due to the increased thrombin generation as gestation progresses resulting in greater fibrin formation (i.e. increased substrate for fibrinolysis).
All the above changes in the hemostatic system normalize to the nonpregnant state by 6–8 weeks post partum.
Thrombophilias are acquired or inherited predispositions to developing venous thrombosis (deep vein thrombosis or pulmonary embolism). These prevalent conditions may have given women a selective evolutionary advantage over nonthrombophilic women by allowing them to cope better with the hemostatic challenge of delivery. For example, it has been demonstrated that women with factor V Leiden (see below) have a reduced risk of intrapartum bleeding compared to normal women [6]. However, in the present day, with modern obstetric practice, this advantage is likely less important than in the past. The downside is that thrombophilias predispose women to venous thrombosis in pregnancy and, as reviewed below, may increase their risk of developing the placenta-mediated pregnancy complications (intrauterine growth restriction (IUGR), pregnancy loss, pre-eclampsia and placental abruption).
Thrombophilias can be acquired or inherited. The known acquired (or partly acquired) thrombophilias include hyperhomocysteinemia, elevated levels of factor VIII and the antiphospholipid antibodies. The known inherited thrombophilias are factor V Leiden (FVL), prothrombin gene variant (PGV), protein C, protein S and antithrombin deficiencies. These inherited thrombophilias are inherited in an autosomal dominant fashion. FVL and PGV are each the result of a single specific mutation, unlike the protein deficiencies which can be caused by multiple different mutations.
Epidemiology
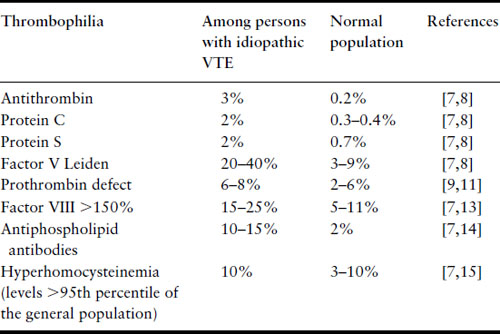
Thrombophilias are common, with a combined general population prevalence of the known thrombophilias over 15% (Table 4.2) [7–15]. Given that they are so common, the obstetric care provider will frequently encounter pregnant patients with thrombophilia. The prevalence of thrombophilias varies greatly in populations of different ethnicity [16]. The most common thrombophilias in Caucasian populations are FVL and PGV. However, in one study, while the carrier frequency of FVL was 5.27% in 2468 Caucasian Americans, the prevalence was much lower in other ethnic populations (2.21% in 407 Hispanic Americans, 1.25% in 80 Native Americans, 1.23% in 650 African Americans, and 0.45% in 442 Asian Americans) [16].
Table 4.2 Prevalence of inherited and acquired thrombophilias
Diagnosis and pathophysiology
Our knowledge about thrombophilia has grown enormously in the past decade; however, because thrombophilia testing is expensive, it is recommended that screening be directed at specific populations where the yield of thrombophilia testing will be higher than for an unselected population (Box 4.1). All women with a previous venous thromboembolism (VTE) who are contemplating pregnancy should be tested for thrombophilia as this will permit better risk stratification. Screening should also be considered in women with a positive family history for thrombophilia or thrombosis (i.e. two or more first-degree relatives affected by thrombosis) where the prevalence of thrombophilia is likely to be increased. A history of placenta-mediated pregnancy complications may be as important as family history when the likelihood of an inherited or acquired thrombophilia is being considered.
The recommended panel of thrombophilia screening tests is presented in Box 4.2. In asymptomatic women with a positive family history for thrombophilia, testing can be limited to the specific thrombophilia identified in the first-degree relative.
Box 4.1 Indications for diagnostic evaluation for thrombophilia
Non-obstetric
- Family history of thrombophilia
- Family history with two or more first-degree relatives with thromboembolism
- History of venous thromboembolism
Obstetric
- Early-onset severe pre-eclampsia <34 wks
- Recurrent early pregnancy loss (2 or more losses less than 10 weeks)
- Unexplained fetal demise >10 wks
- Intrauterine growth restriction <10th percentile adjusted for gender and gestational age
- Placental abruption leading to delivery
Box 4.2 Thrombophilias to be screened for in patients with an indication for thrombophilia testing (see also Table 3.4)
- Factor V Leiden (FVL)
- PCR for prothrombin gene mutation G20210A (PGV)
- Protein S deficiency (PS)
- Protein C deficiency (PC)
- Antithrombin deficiency (AT)
- Antiphospholipid antibodies
- Anticardiolipin antibodies (ACLA), anti-beta-2 glycoprotein I antibodies and lupus anticoagulant (LA)
- Hyperhomocysteinemia (HCY)
A careful history and physical exam should be performed on all pregnant women suspected of having a thrombophilia. Any history suggestive of thrombophilia should be elicited, including a personal history of venous thrombosis, recurrent thrombosis or thrombosis at an unusual site. A personal history of arterial thrombosis and previous pregnancy complications are also pertinent. The presence of a family history of venous thrombosis or thrombophilia should be determined. Patients with a personal and family history of thrombosis often have more than one thrombophilic state and therefore should be tested for all thrombophilias.
Diagnostic tests for thrombophilia and the effects of pregnancy on these tests are discussed in detail below. The definitive diagnosis of thrombophilia can only be made by laboratory testing in a laboratory that is skilled at handling such requests. When a reliable laboratory is not available, the managing physician should refer patients to a thrombosis specialist. A specialist should also be consulted when the thrombophilia test results are confusing, ambiguous or equivocal.
Activated protein C resistance and factor V Leiden
Activated protein C (APC) is an important element in the coagulation cascade, in which it functions to suppress activated coagulation. APC, with its co-factors protein S and factor V, forms a complex that dampens coagulation through proteolytic action on factor Va and factor VIIIa [17–19]. APC resistance (APCR) results in less dampening of activated coagulation, leading to an increased propensity for abnormal thrombosis. APCR in 90% of cases is due to a mutation referred to as factor V Leiden (FVL) after the city in The Netherlands where it was initially described. FVL is a a single missense mutation in the factor V gene that renders the gene resistant to cleavage and inactivation by APC.
Factor V Leiden should be diagnosed in a laboratory with the appropriate expertise. The following approach should be considered.
- Measure the resistance to activated protein C (PCR) in plasma diluted with FV-deficient plasma as an initial screening test. FV-deficient APCR assays for FVL are highly sensitive and specific [20] and are unmodified by pregnancy [2], oral anticoagulants [21] or factor VIII levels [21]. FV-deficient APCR assays are less expensive to perform than DNA-based PCR assays but may not be readily available in all labs. Other assays may show increased resistance to AP C during pregnancy that will not be present in the nonpregnant state and therefore should not be used.
- Confirm the diagnosis (or if the FV-deficient assay is not available, begin your investigations during pregnancy) with DNA testing using direct PCR testing to determine hetero- or homozygous state and exclude false positives. False positives secondary to antiphospholipid antibodies can occur so all positive APCR tests should be confirmed with DNA testing.
Prothrombin gene variant
Prothrombin, the precursor to thrombin, is encoded by a gene on the long arm of chromosome 11. In 1996, a mutation in the prothrombin gene, called G20210A or PGV, was identified which results in a single base substitution from guanine to adenosine on the 3’ untranslated end at nucleotide 20210 [11]. PGV results in increased prothrombin levels and a predisposition to venous thromboembolic disease [22].
Prothrombin gene variant should be diagnosed in a laboratory with the appropriate expertise and should be tested with DNA-based PCR testing.
Protein C, protein S and antithrombin deficiencies
These anticoagulant proteins all suppress activated coagulation and deficiency results in increased risk of venous thrombosis, with lifetime VTE risk in the 50–60% range. These deficiencies, like FVL and PGV, have autosomal-dominant inheritance, with variable penetrance (i.e. they are tendencies to develop VTE, not certainties). However, unlike the unique mutations that lead to FVL and PGV, hundreds of rare insertions, deletions and point mutations account for these protein deficiencies. Patients with antithrombin deficiency seem to have the highest risk for thrombosis (~65% lifetime risk).
Protein C should be tested in an accredited lab with the appropriate expertise. The following approach should be considered and pitfalls avoided.
- Recognize that at least two abnormally low results (and no normal results) are required to make the diagnosis of protein C deficiency.
- Perform a functional assay (preferably an amidolytic assay) as a screening test. Antigenic assays help distinguish between quantitative and qualitative deficiencies, but the vast majority of patients have both low activity and low antigen levels. Distinguishing between quantitative and qualitative deficiencies is of little clinical value and a functional assay is the appropriate test to begin with.
- Avoid testing patients on oral anticoagulants as protein C levels decrease with oral anticoagulants [23]. Protein C levels can also decrease with acute thrombosis [24], liver disease [25], vitamin K deficiency and sepsis [25]. Protein C levels are not influenced by pregnancy [2]. False positives secondary to APCR and elevated FVIII levels can be minimized by using amidolytic assays with snake venom as the activator [26].
Protein S should be tested in an accredited lab with the appropriate expertise. The following approach should be considered and pitfalls avoided.
- At least two abnormally low results (and no normal results) are required to make the diagnosis of protein S deficiency.
- Perform antigenic assays for free protein S when screening for protein S deficiency. Recognize that the antigenic assay should be a free protein S assay. Free protein S levels are more sensitive and specific than total protein S levels in patients with genotypically confirmed protein S deficiency [27,28].
- Functional assays are not specific or sensitive for protein S deficiency and should not be used to diagnose protein S deficiency [29].
- Avoid testing patients on oral anticoagulants [23], oral contraceptives, hormone replacement therapy or during pregnancy [2] or acute thrombotic events [24], which can all lead to decreased protein S levels. Protein S levels are also decreased by liver disease, vitamin K deficiency, disseminated intravascular coagulation (DIC) and nephrotic syndrome [25,30]. Functional protein S assays can be falsely low in patients with APCR [31].
Antithrombin deficiency should be diagnosed in a lab with the appropriate expertise. The following approach should be considered and pitfalls avoided.
- Recognize that at least two abnormally low results (and no normal results) are required to make the diagnosis.
- Perform a heparin co-factor activity-based functional assay. These assays can detect all cases of clinically relevant antithrombin (AT) deficiency [32]. Patients with AT deficiency may have low activity and low antigen levels or low activity and normal antigen levels. Hence, functional testing is adequate to detect all relevant AT deficiency.
- Avoid testing patients on oral anticoagulants or heparin therapy; false-positive results are common with heparin therapy [33]. False-negative results can result from vitamin K antagonist therapy [34]. False-positive results (low levels) can also be seen with pre-eclampsia, liver disease, DIC, nephrotic syndrome and acute thrombosis [25].
- Acquired antithrombin deficiency may occur with nephrotic syndrome or pre-eclampsia.
Antiphospholipid antibodies
Antiphospholipid antibodies predispose to venous and arterial thrombosis and recurrent miscarriage. The exact mechanism by which antiphospholipid antibodies (lupus anticoagulants, anti-beta-2 glycoprotein I antibodies and anticardiolipin antibodies (ACLA)) predispose to thrombosis or miscarriage is unknown.
Antiphospholipid antibody testing is complex and confusing in the best of hands. The lab diagnosis of antiphospholipid antibody syndrome is complicated by the lack of specific tests, the heterogeneity of the antibodies and problems with test standardization. While the following approach and pitfalls must be considered, if results are ambiguous a thrombosis expert should be consulted.
- Perform anticardiolipin and anti-beta-2 glycoprotein I antibody testing (IgG and IgM) by enzyme-linked immunosorbent assay (ELISA). Test for lupus anticoagulant (LA) activity with a phospholipid-dependent clotting assay (activated partial thromboplastin time (aPTT), kaolin clotting time (KCT), dilute Russell’s viper venom time (DRVVT), dilute prothrombin time). LA is diagnosed if the following criteria are met: one or more phospholipid-dependent test(s) is prolonged; the above prolongation is not corrected when patient and healthy control plasma are mixed (i.e. do not correct with a mixing study); the prolongation for patient plasma is corrected by adding phospholipid in the test system (platelet phospholipid neutralization) [35]. Testing for other antiphospholipid antibodies is not usually necessary as they are far less likely to be of clinical significance.
- Recognize that a “positive” result requires at least one abnormal result but that persistently positive results, higher titers (for ACLA) or greater prolongation of phospholipid-dependent tests (for LA) are more specific [36]. Positive anticardiolipin results can be transient and may be related to acute infection or other illness. Mild elevations of ACLA antibodies are nonspecific and do not necessarily indicate a hypercoagulable state. Positive results should be confirmed over time to ensure that they are not transient [37].
Hyperhomocysteinemia
Hyperhomocysteinemia is a common cause of hypercoagulability and a risk factor for VTE and arterial thrombosis. Homocysteine levels are usually maintained within a narrow range of concentrations via a complex series of enzymes and co-factors. These include folic acid, vitamin B12 and vitamin B6. It has long been known that the severe form of hyperhomocysteinemia, an inborn error of metabolism, causes both arterial and venous thrombosis. However, more recently it was discovered that mild acquired hyperhomocysteinemia, at times resulting from folic acid, vitamin B6 or vitamin B12 deficiency, renal failure or certain drugs, was also associated with an increased risk for thrombosis [15].
The common C677T mutation (and the less common a1A298C mutation) for methylenetetra-hydrofolate reductase (MTHFR) is associated with hyperhomocysteinemia; however, its correlation with VTE remains controversial. Meta-analyses have failed to find a strong association between VTE and homozygosity for MTHFR C677T [38]. Similarly, this mutation has not been clearly implicated in placenta-mediated pregnancy complications (see Table 4.2). Homocysteine levels decrease in pregnancy, probably related to increased circulating volume, decreased albumin levels and the use of prenatal vitamins containing folic acid [39].
The following diagnostic approach and pitfalls should be considered.
- Perform fasting homocysteine levels as they are simpler than and as reliable as postmethionine loading tests [40]. The risk of VTE and cardiovascular disease begins to increase as fasting plasma homocysteine levels exceed 10 μmol/L. Do not test for the MTHFR gene mutation (MTHFR) as the association appears to be with homocysteine and not MTHFR.
- Plasma homocysteine levels increase with age, renal impairment and low dietary B6, B12 and folate intake [40]. If hyperhomocysteinemia is diagnosed, exclude B12 and folate deficiency by testing these levels as other important underlying diseases need to be excluded (e.g. celiac disease, pernicious anemia).
Elevated factor VIII
Elevated FVIII levels have been identified as a risk factor for a first VTE and a predictor of recurrent VTE in those that have had VTE. However, the appropriate upper cut-off level for defining a FVIII thrombophilia is debated and inter-reagent and intermachine variability is very high, currently limiting the clinical utility of elevated FVIII levels and confidence in establishing or excluding this thrombophilia [41]. Also, it is well recognized (see above) that FVIII levels increase in pregnancy, further hampering the ability to make an accurate diagnosis of pathologically elevated FVIII levels. Furthermore, elevated FVIII levels and association with placenta-mediated pregnancy complications have received little attention in the literature thus far and we do not recommend this testing during pregnancy at the time of writing.
Clinical presentations
Thrombophilia as a predisposing factor for VTE in pregnancy
Pregnancy and thrombophilia act as synergistic risks and increase the risk of thrombosis during pregnancy. Case–control evidence indicates that women with thrombophilia have a 3–15-fold higher risk of VTE during pregnancy [22,42,43]. Although asymptomatic thrombophilic women (i.e. no prior VTE) are at greater relative risk of developing VTE, what is known about absolute risks of antepartum VTE is reassuring, with absolute risks of VTE in pregnant thrombophilic women being low with the common thrombophilias (e.g. ~1% with FVL) [44,45]. These absolute risks are poorly defined with the less common and more potent thrombophilias (e.g. AT deficiency) because large prospective cohort studies have not been conducted.
In women with previous VTE (proximal deep vein thrombosis (DVT) or pulmonary embolism (PE)) who are no longer on anticoagulants, the absolute risks are more of a concern. In a study of 125 untreated women with a single previous VTE, 1/10 (10%, 95% confidence interval (CI) 0.3–44%) women with a prior unprovoked VTE and thrombophilia and 2/21(10%, 95% CI 1–30%) women with a prior VTE and an identifiable thrombophilia had antepartum recurrences [46]. These women had a risk of postpartum recurrence of 2.4% (3/125) (95% CI 0.5–7.0%) despite all study participants being recommended postpartum prophylaxis, demonstrating the even higher risk during this brief period when delivery and immobilization are added to the synergistic risks of thrombophilia and the hypercoagulability of pregnancy. In women with combined thrombophilic defects, homozygosity for genetic thrombophilias or type 1 antithrombin deficiency (abnormal functional and antigenic levels of antithrombin), absolute risks have been reported to be as high as 10–35% [47].
Thrombophilia as a predisposing factor for placenta-mediated pregnancy complications
Venous thromboembolism is the most common cause of maternal mortality in developed countries but placenta-mediated pregnancy complications are also significant causes of maternal and fetal mortality and morbidity. These complications include IUGR, pre-eclampsia, and pregnancy loss (embryonic and fetal loss and placental abruption) [48–50]. The placenta-mediated pregnancy complications, like thrombophilias, are common, occurring in over 1/6 pregnancies, and are important conditions with a large impact on maternal and fetal health. Pre-eclampsia is a frequent cause of fetal and neonatal morbidity and mortality [51]. IUGR is associated with increased risk for fetal demise and may result in long-term effects in children, including developmental delay and poor school performance. Those afflicted by IUGR are also significantly less likely to attain higher academic and professional achievement as adults [52]. Pregnancy loss is a devastating event for pregnant women and their families.
A successful pregnancy demands the development of adequate placental circulation. The placenta-mediated pregnancy complications are thought to be due to inadequate placenta circulation (in part due to thrombosis) [53–56] and/or abnormal placentation (which is likely influenced by coagulation activation and fibrinolysis at the maternal/fetal interface) [57,58]. While all placenta-related complications are likely multifactorial, and most happen in the absence of a thrombophilia, it is biologically plausible that thrombophilia may increase the risk of both placental insufficiency due to placental micro- and/or macrovascular thrombosis and abnormal placentation due to alterations in coagulation and fibrinolysis at the maternal/fetal interface.
Factor V Leiden and PGV are the best studied thrombophilias to date (due to their high prevalence and the ability to simply and reliably test for them in large populations with PCR assays). As FVL and PGV are the least potent of the thrombophilias, it should follow that if they cause the placenta-mediated pregnancy complications then the more potent thrombophilias may also cause these complications. Similarly, if it is demonstrated that anticoagulants reduce the risk of placenta-mediated pregnancy complications in FVL or PGV pregnancies then it is likely that they will be beneficial in women with more potent thrombophilias.
Thrombophilia and placenta-mediated pregnancy complications: a causality assessment
To begin a causality assessment, statistical association in case–control or cohort studies is required. While statistical association may be a matter of fact, causation is a matter of opinion. Statistical association is but one criterion that is used to establish causation. As in criminal courts, a case for causal association is developed and opinion rendered by expert interpreters but ultimately clinicians must be judge and jury and determine for themselves whether causation has been proven. As detailed below, we submit that the case for thrombophilias causing placenta-mediated complications is at best weak and does not yet meet the criteria to establish causality.
The definitions of causation [59] and criteria for causality assessment are hotly debated in epidemiologic literature but Hill’s criteria [60] are widely used and have withstood the test of time. Hill’s criteria include strength of association, consistency of association, temporal relationship, specificity, biologic plausibility, biologic gradient, coherence, analogy, and experimentation.
- Strength of association: this criterion states that if factor X causes disease Y then variation of factor X will influence the probability of disease Y [61]. Furthermore, if X and Y are strongly linked then the case for causation is improved. The statistical association between factor X and disease Y can be studied in observational studies (case–control and cohort studies). In case–control studies, the prevalence of factor X is measured in those who did and did not develop disease Y. In cohort studies, those with and without factor X are followed over time to see who develops disease Y. Meta-analyses of observational studies suggest that women with thrombophilia are at increased risk of pre-eclampsia (see Table 4.2) [62,63], IUGR [63,64], abruptio placentae [65], miscarriage, and stillbirth [63,66]. However, on close inspection of these meta-analyses where the confidence intervals are narrow (i.e. where there are a larger number of events available for study), the point estimates of the odds ratios (effect sizes) are small (1.5–4 range), suggesting that the association is weak. To put these absolute odds ratios in context, odds ratios in the 1000 range are seen in single-gene diseases like hemochromatosis [67]. The prospective cohort studies conducted to date have suggested no association and have been large enough to exclude relative risks >4 (see below).
- Consistency of association: this criterion states that if an observed effect of factor X on disease Y is consistently shown in studies of different designs and in different populations then causation is more likely [61]. While the meta-analyses and systematic reviews of case–control studies have overall suggested an association, many individual studies have not (see Table 4.2). To be adequately powered (>80%) to detect an odds ratio of 2 or greater with a risk factor that has a prevalence of less than 5%, a sample size of greater than 1000 is required in a 1:1 case–control study. With but one exception [68], all the case–control studies published to date have been smaller than 1000 patients. It follows that the inconsistency seen in the case–control studies published to date may be due to small actual effect sizes combined with insufficient power (i.e. type II error). If we turn our attention to other study designs, as introduced above, none of the published prospective cohort studies examining the association between thrombophilia and the placenta-mediated pregnancy complications have demonstrated a statistical association. However, if an increase in relative risk truly exists, and the increase is of the order of twofold, then the individual prospective cohort studies have been underpowered.
- Temporal relationship: this criterion is considered by definition necessary and states that exposure to risk factor X should precede disease Y. Clearly, this criterion is met for the inherited thrombophilias, but for elevated FVIII, hyperhomocysteinemia and the antiphospholipid antibodies, this criterion has not been necessarily met in investigations conducted to date.
- Specificity

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


