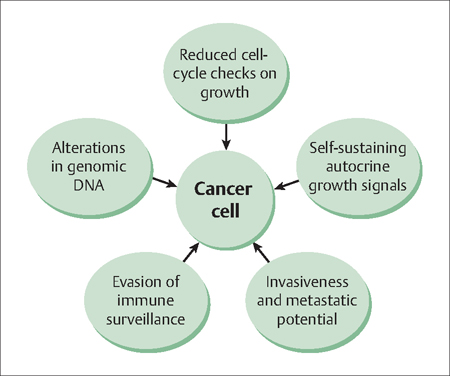
45 The Biology of Cancer Robert L. Barbieri Two characteristic features of cancer cells are unrestrained proliferation and invasion into adjacent structures (Fig. 45.1). The mechanisms by which normal adult cells develop the capacity for both unrestrained proliferation and invasiveness are not full characterized, but altered function of genomic DNA is critical to the pheno-type. Genomic DNA can be permanently altered by many mechanisms. Relevant to gynecological disease, the biological mechanisms that can alter the function of genomic DNA include viral infection (cervical cancer), excess hormonal stimulation (endometrial cancer), and various mutations that reduce the cell’s capacity for DNA repair or that disrupt cell cycle control checkpoints(endometrial and ovarian cancer). These mechanisms are explored in this chapter, using examples from gynecologic cancer. In 1983, zur Hausen reported the presence of human papilloma virus (HPV) types 16 and 18 in cervical cancer biopsies. Subsequent research led to an understanding of the molecular mechanisms by which infection with HPV-16 and HPV-18, if left untreated, cause cervical cancer. HPV type 16 and 18 infections have also been shown to cause cancer of the vulva, vagina, penis, anus, and mouth. The circle of medical science, from molecular discovery to effective patient and population treatment, was closed in 2005 when a vaccine for HPV-16 and HPV-18 to combat cancer became widely available. In 2008, zur Hausen received the Nobel Prize in Medicine for this important discovery. Zur Hausen has gone on to discover that polyomaviruses can cause skin cancer. The role of viruses in the etiology of cancer continues to evolve. HPV infection is endemic among sexually active men and women. There are over 115 types of HPV infection and over 40 types are specific for the anogenital epithelium. In most cases, infection with HPV results in latent infection without symptoms, physical findings, or cellular changes in the cervix. In some cases, HPV infection results in active infection 2–8 months after initial exposure, which results in cellular changes in the cervix including nuclear enlargement, multinucleation, and perinuclear cytoplasmic clearing. These cellular findings are often diagnosed on cervical cytology as low-grade squamous intra-epithelial neoplasia. Immunological response to the infection may result in resolution or chronic infection. If the HPV integrates into the cell genome, high-grade squamous intra-epithelial lesions and cancer may result. The HPV types that are most likely to result in genomic integration and cancer are 16, 18, 45, 59, and 35. Integration of HPV into the genome results in overexpression of E6 and E7, two HPV proteins that release the cell from cell cycle checkpoints. Fig. 45.1 Characteristics and behavior of cancer cells The viral E6 protein binds to the tumor suppressor gene p53 (which blocks the progression of cells through the late G1 phase), reducing its activity and increasing its degradation. This reduction in expression of p53 releases the cell from the G1 cell-cycle checkpoint, and permits the cell to replicate more rapidly. In addition, normal levels of p53 activity prevent cells with DNA damage from proliferating. Reduction in p53 activity is associated with chromosome instability, increasing the likelihood that additional detrimental mutations will accrue. The viral E7 protein binds to the retinoblastoma protein (Rb) which causes the release and activation of transcription factor E2F, which promotes cell proliferation. In addition, when E7 is released by infected cells it causes an increase in the production of interleukins 6 and 8, which in turn increases the rate of cell proliferation. The ability of certain HPV types to integrate into the genome, over-express E6 and E7, and thereby decrease the function of two important tumor suppressor proteins, p53 and Rb, is central to the oncogenic potential of these types. It is likely that the double-hit on two important tumor suppressor networks, p53 and Rb, is critical to evading normal cell mechanisms for restraining growth. Estradiol stimulates endometrial cell proliferation. Progesterone blocks estradiol stimulation of endometrial cells. In the high concentrations observed in pregnancy, progesterone can cause terminal differentiation of endometrial cells to form decidua. These cells have markedly reduced capacity for further proliferation. The essence of the estradiol-induced proliferation of the endometrium and the progesterone-induced secretory differentiation and block of further proliferation represents the ebb—flow of the endometrium during the menstrual cycle. Estradiol causes the endometrium to grow, progesterone stops the growth and prepares (by differentiation) the endometrium for implantation. At menses, withdrawal of both the estradiol and progesterone disrupts the integrity of the endometrium and results in menstrual bleeding. With the next cycle, increase in ovarian production of estradiol causes the endometrium to stabilize, start growing, and stop bleeding. Constant estradiol stimulation of the endometrium results in the development of type I, endometrioid, endometrial cancer. These lesions have characteristic mutations in the PTEN, k-ras and beta-catenin genes and demonstrate DNA microsatellite instability. The PTEN (phosphatase and tensin homologue) gene is a suppressor of estrogen-stimulated endometrial cell growth. Mutations in the PTEN gene that result in loss of function cause greater endometrial cell growth in response to a given concentration (dose) of estradiol. In addition, endometrial cancer cells appear to sometimes amplify the estrogen receptor gene, resulting in overexpression of active estrogen receptor, further sensitizing the cell to the growth-promoting effects of estradiol stimulation.
Cervical Cancer is Caused by the Human Papilloma Virus
Endometrial Cancer is Caused by Unopposed Estrogen Stimulation
Endometrial and Ovarian Cancers are Caused by Germ-Line Mutations in DNA Mismatch Repair Genes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


