Structure of a chromosome
The first 22 pairs of chromosomes are known as autosomes and have been designated the numbers 1 through to 22 depending upon the decreasing relative length of the chromosome pairs. The composition of the 23rd pair of chromosomes, the sex chromosomes, determines a person’s gender with female individuals having two X chromosomes and male individuals having an X and a Y chromosome.
In the creation of germ cells, the number of chromosomes is halved so that each ovum and spermatozoon contains only one chromosome from each pair. Germ cells therefore have 23 chromosomes, known as the haploid number, whereas somatic cells have 46 chromosomes, referred to as the diploid number.
Genes
Each chromosome is composed of tightly coiled threads of DNA organised around proteins called histones (Figure 18.1). A gene is a specific sequence of DNA that carries the instructions for the construction of a protein (see Chapter 5 for further information on the structure of DNA and protein synthesis). The human genome is estimated to contain approximately 20 000 genes.
Cell Division
Mitosis
When somatic cells divide, they go through a process known as mitosis, whereby the cell duplicates its DNA and cleaves to generate two identical daughter cells also with 23 pairs, or the diploid number, of chromosomes (Figure 18.2).
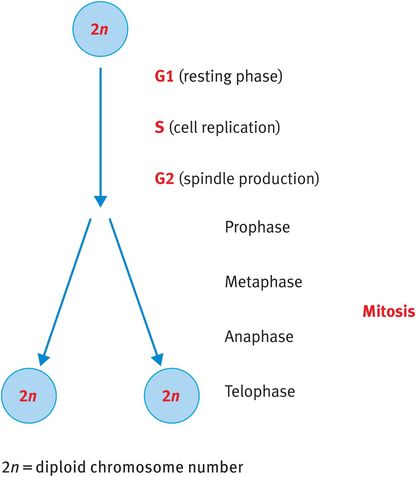
Mitosis
Meiosis
By contrast, when germ cells divide to produce ova or sperm, they undergo meiosis. Meiosis is unique to the germ cells and is essential to reduce the somatic diploid number of 46 chromosomes to the haploid number of 23 chromosomes (Figure 18.3). This reduction ensures that, when fertilisation occurs, the zygote has 46 chromosomes rather than 92, which would be the result of fertilisation involving two diploid germ cells.
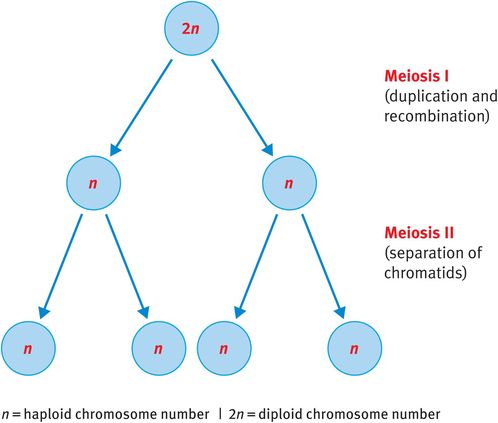
Meiosis
Meiosis consists of two consecutive division cycles that are designated meiosis I and meiosis II.
Meiosis I
Chromosomes duplicate at the beginning of meiosis I. Homologous chromosomes, now consisting of identical sister chromatids, align and reciprocal exchange of genetic material occurs between homologous but nonidentical chromatids. This process, known as recombination, enables the mixing of paternally and maternally inherited genes, ensuring that children inherit unique gene combinations. Following recombination, the pairs of duplicated chromosomes align at the spindle equator and one duplicated chromosome from each pair, consisting of two sister chromatids, is pulled to either pole of the cell before cytokinesis generates two daughter cells. Therefore, at the end of meiosis I each daughter cell has 23 chromosomes, each consisting of two chromatids (Figure 18.3).
Meiosis Ii
The phases of meiosis II resemble those of mitosis except that, as each chromosome is already duplicated at the beginning of meiosis II, there is no requirement for additional replication. Therefore, the single chromosomes align at the spindle equator, the spindle matrix is generated and sister chromatids are pulled to opposite poles of the cell before cytokinesis.
There are 46 chromosomes (23 pairs) in each somatic cell; this is the diploid number.
There are 23 individual chromosomes, one from each chromosome pair, in the ova and sperm; this is the haploid number.
The chromosomes contain approximately 20 000 genes.
Genes code for the production of specific proteins.
Mitosis is the process whereby somatic cells divide. The progenitor cell has 46 chromosomes and each daughter cell has 46 chromosomes.
Meiosis occurs in the generation of germ cells. It has two distinct phases:
During meiosis I, chromosomes duplicate, paternally and maternally inherited chromosomes are recombined and the diploid number is reduced to the haploid number.
During meiosis II, splitting of sister chromatids occurs.
Therefore, by the end of meiosis, four daughter cells are generated from one dividing germ cell, paternal and maternal genetic material is mixed and each germ cell has only 23 chromosomes.
Thus, by the end of meiosis, four daughter cells are generated from each dividing germ cell. Each of these daughter cells should have 23 individual chromosomes, representing one chromosome from each pair of autosomes in addition to an X chromosome in ova and either an X chromosome or a Y chromosome in sperm. As a result of recombination during meiosis I, each germ cell will have a unique combination of both paternally and maternally inherited genes.
Of note, in female meiosis, three of the daughter cells constitute polar bodies and will not survive; only one daughter cell becomes an ovum. By contrast, all four daughter cells produced from male meiosis develop into spermatozoa.
Chromosome abnormalities
An abnormality of chromosome number is known as aneuploidy. Aneuploidy can result from various mechanisms, including nondisjunction and translocation.
Nondisjunction
Occasionally, pairs of homologous chromosomes at meiosis I or sister chromatids during mitosis anaphase do not separate and so both chromosomes are passed to one daughter cell and none to the other. One daughter cell therefore has an additional chromosome, a phenomenon known as trisomy, whereas the other is missing one chromosome, known as monosomy.
Common trisomic conditions include Down syndrome (trisomy 21), Edward’s syndrome (trisomy 18), Patau syndrome (trisomy 13) and Klinefelter syndrome (47,XXY); Table 18.1 provides an overview of these conditions. Turner syndrome (45,X) is the most common monosomy identified at birth. In addition, mosaic (where only a proportion of cells have the additional chromosome) trisomy 16 and mosaic trisomy 8 are occasionally identified postnatally. Other trisomies and monosomies are rarely seen postnatally as they are not compatible with term development of the fetus.
| Syndrome name | Additional chromosome | Clinical features |
|---|---|---|
| Down syndrome | 21 | Moderate to severe learning difficulties (100%) Cardiac problems (40–50%, VSD, ASD, AVSD) Hypothyroidism (20–40%) Dementia (10–15%) Acute lymphoblastic leukaemia |
| Edwards syndrome | 18 | Profound learning difficulties (100%) Congenital heart disease (90%, commonly VSD) Facial clefts Spina bifida Clenched hands Rocker-bottom feet |
| Patau syndrome | 13 | Profound learning difficulties (100%) Congenital heart disease (80%, VSD, ASD) Holoprosencephaly (60–70%) Microphthalmia/anophthalmia (60–70%) Postaxial polydactyly (60–70%) Scalp defects Cleft lip/palate (60–70%) Omphalocele Renal anomalies |
| Klinefelter syndrome | X (in an XY individual) | Slightly decreased IQ but within the normal range Tall stature Infertility Transient gynaecomastia |
| Triple X syndrome | X (in an XX individual) | Slightly decreased IQ but within the normal range Tall stature Normal fertility |
| XYY syndrome | Y (in an XY individual) | Slightly decreased IQ but within the normal range Tall stature after puberty Behavioural problems |
ASD = atrial septal defect | AVSD = atrioventricular septal defect | VSD = ventricular septal defect
The risk of chromosome nondisjunction increases with advancing maternal age (Table 18.2).
| Age (years) | Risk |
|---|---|
| 20 | 1 in 1500 |
| 30 | 1 in 900 |
| 34 | 1 in 500 |
| 36 | 1 in 300 |
| 38 | 1 in 200 |
| 40 | 1 in 100 |
| 42 | 1 in 60 |
| 45 | 1 in 30 |
Down syndrome
Nondisjunction resulting in three separate copies of chromosome 21 accounts for more than 95% of the total incidence of Down syndrome. Alternative causes include Robertsonian translocations and other chromosome rearrangements.
All individuals with Down syndrome have some degree of learning difficulties, usually moderate. In addition, atrioventricular septal defect is 1000 times more common in children with Down syndrome than in the general population. The facial appearance of children and adults with Down syndrome is characteristic and consists of upslanting palpebral fissures, epicanthic folds (folds of skin from the upper eyelid continue down to the nasal corner), a flat midface and brachycephaly (short anterior–posterior diameter of the cranium). Other associated features are shown in Table 18.1.
Edwards syndrome
Edwards syndrome is associated with a poor prenatal and postnatal outcome. The great majority of occurrences are caused by meiotic nondisjunction but, as with Down syndrome, the syndrome can be attributable to unbalanced Robertsonian translocations and other chromosome rearrangements.
Edwards syndrome is usually detected prenatally either through aneuploidy screening methods or through identification of a combination of many associated prenatal anomalies (Table 18.1). Babies with Edwards syndrome who survive to term are likely to die within the first year of life (usually within the first months). Very rarely, children have survived beyond the first year with profound cognitive impairment and associated anomalies described in Table 18.1.
Patau syndrome
Patau syndrome, caused by an additional copy of chromosome 13, is also associated with a high rate of spontaneous pregnancy loss and, if the baby survives to term, poor postnatal outcome. Most (90%) occurrences of Patau syndrome are caused by nondisjunction of chromosome 13 (the vast majority of which occur during maternal meiosis). The remainder are attributable to unbalanced translocations (primarily Robertsonian translocations).
Klinefelter syndrome
Individuals with Klinefelter syndrome have an additional sex chromosome, with a karyotype of 47,XXY. Many individuals with Klinefelter syndrome are not diagnosed unless as an incidental finding prenatally during chorionic villus sampling (CVS) or amniocentesis or in adulthood during infertility investigations. As Klinefelter syndrome does not, in general, cause an increase in nuchal translucency (NT) thickness and does not result in features that are evident on the second trimester anomaly scan, the condition is usually not identified prenatally.
Other sex chromosome abnormalities resulting from nondisjunction
These are illustrated in Table 18.1. The most common examples are a 47,XXX karyotype (triple X syndrome) in female persons and a 47,XYY karyotype in male persons. Triple X syndrome is usually identified incidentally as it causes few problems prenatally or postnatally.
Turner syndrome
In general, girls with Turner syndrome (45,X) have IQ scores 10–15 points lower than their siblings but they attend mainstream school and do not have learning difficulties. Many fetuses with 45,X do not survive to term because of fetal hydrops. Indeed, Turner syndrome should be considered in any fetus with an NT thickness above 4 mm. The attrition rate for conceptions with Turner syndrome is reported to be very high, so if the syndrome is diagnosed in the first trimester, parents should be advised of a high risk of spontaneous miscarriage.
Translocations
Reciprocal translocations
Chromosome rearrangements involving the transfer of genetic material between two nonhomologous chromosomes are known as reciprocal translocations (Figure 18.4). The most common reciprocal translocation in humans, t(11; 22)(q 23; q 11), involves chromosomes 11 and 22.
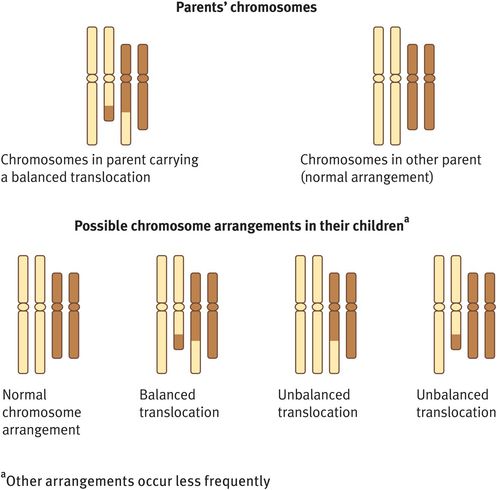
Reciprocal translocation
Reciprocal translocations can be either balanced or unbalanced. In general, a balanced exchange of chromosome material does not have phenotypic consequences as the adult or child has a normal amount of genetic material. However, if unbalanced, there will be one chromosome with missing genetic material (a monosomy) and another chromosome with duplicated genetic material (a trisomy), a combination that is usually associated with developmental and other congenital problems.
If a woman with a balanced reciprocal translocation is seen before she conceives, she should be advised that a pregnancy can have four possible outcomes (Figure 18.4):
A normal pregnancy with a normal karyotype.
A normal pregnancy with the familial balanced reciprocal translocation.
A spontaneous miscarriage owing to an unbalanced product of the familial reciprocal translocation.
A pregnancy that goes to term but the child has a high likelihood of learning difficulties and possibly other congenital anomalies associated with an unbalanced product of the familial reciprocal translocation.
The relative proportion of pregnancies that spontaneously miscarry or go to term with problems depends upon the chromosomes involved and the size of the translocated chromosome segments.
If an unbalanced reciprocal translocation is detected prenatally, it is likely to be associated with fetal anomalies and the parents should be counselled accordingly. If a balanced reciprocal translocation is identified, the parents should be karyotyped to determine whether it is de novo. If it is familial and the parent carrying the balanced rearrangement is healthy with no learning or other medical difficulties, the baby is unlikely to have problems related to the translocation. However, if the balanced translocation has arisen for the first time in the fetus (it is de novo), it may or may not be associated with problems and it would be recommended to undertake an array comparative genomic hybridisation (aCGH) to investigate for a microdeletion.
Robertsonian translocations
A Robertsonian translocation is a chromosome rearrangement that involves fusion of the long arms of two acrocentric chromosomes and loss of their short arms (Figure 18.5). The genes contained on the short arms are represented elsewhere and so their loss does not result in any phenotypic effect of acrocentric chromosomes.
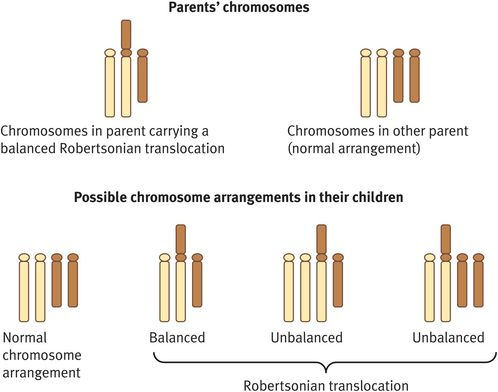
Robertsonian translocation
By definition, Robertsonian translocations only involve the five acrocentric chromosomes 13, 14, 15, 21 and 22. The most common Robertsonian translocations are rob(13q; 14q), which involves fusion of chromosome 13 to chromosome 14, and rob(14q; 21q), which involves fusion of chromosome 14 to chromosome 21. Occasionally, the long arms of two homologous chromosomes may fuse resulting in an isochromosome.
Individuals who are seen before conception to discuss the implications of a balanced Robertsonian translocation for a future pregnancy should be advised of a number of possible outcomes (Figure 18.5):
A normal pregnancy with a normal karyotype.
A normal pregnancy associated with a balanced Robertsonian translocation.
A spontaneous miscarriage attributable to an unbalanced Robertsonian translocation (the fetus will be trisomic or monosomic for one of the component chromosomes).
A spontaneous miscarriage owing to an imprinting defect.
A liveborn child with abnormalities; in all instances, these will be attributable to trisomy rather than monosomy, as the latter will always result in spontaneous miscarriage; in the common rob(13q; 14q) and rob(14q; 21q) translocations, there is a risk of a child with Patau syndrome or Down syndrome, respectively; these risks are higher if the mother carries the balanced translocation (10–15%) than if the father carries it (below 1%).
A liveborn child with abnormalities associated with an imprinting defect (risk below 0.5%).
Imprinting is the process by which one parental allele is preferentially silenced according to its parental origin. Of the five acrocentric chromosomes, imprinted genes are found only on chromosomes 14 and 15. Imprinting abnormalities associated with Robertsonian translocations are therefore only of concern if one or both of these two chromosomes are involved. Following mitosis, a trisomic conceptus may occur, but this may be ‘corrected’ through trisomy rescue. This reduction of three copies to two copies leaves the cell with an apparently balanced arrangement. However, if both the remaining chromosomes are of the same parental origin (known as uniparental disomy or UPD) and contain imprinted genes, there may be phenotypic consequences.
Babies with maternal UPD14 generally survive to term and are small, have learning difficulties, hypotonia and relative macrocephaly. By contrast, paternal UPD14 tends to result in spontaneous miscarriage, but if babies do survive to term they have severe problems including profound learning difficulties, feeding difficulties and joint contractures.
Paternal UPD15 and maternal UPD15 cause Angelman syndrome and Prader–Willi syndrome, respectively. These are well described syndromes that are both associated with severe learning difficulties. In addition, children with Angelman syndrome have a characteristic facial appearance and an ataxic gait. Children with Prader–Willi syndrome are characteristically poor feeders in the neonatal period but develop an insatiable appetite from childhood onwards. In addition, they may be hypotonic and of short stature.
Chromosome Microdeletions
Chromosome microdeletions are an important cause of a number of different syndromes illustrated in Table 18.3. Microarray aCGH is now the technique of choice for identifying chromosome microdeletions and microduplications. However, not all microdeletions and microduplications are pathogenic: we now know of a number of clearly nonpathogenic deletions/duplications (designated as benign copy number variants) and a number of possible susceptibility loci, the pathogenicity of which is currently unclear.
| Microdeletion | Syndrome name | Cardinal features |
|---|---|---|
| 1p36 | Severe learning difficulties Seizures Hypotonia Feeding difficulties Characteristic facial appearance with low-set horizontal eyebrows and deep-set eyes | |
| 4p15 | Wolf–Hirschhorn | Severe learning difficulties Low birth weight and postnatal failure to thrive Characteristic facial appearance with ‘Greek helmet’ profile |
| 5p15 | Cri du chat | Severe learning difficulties Characteristic cat-like cry Characteristic facial appearance with bitemporal narrowing, hypertelorism and downslanted palpebral fissures |
| 7q11 | Williams | Mild to severe learning difficulties Cardiac problems, particularly supravalvular aortic/pulmonary stenosis Renal artery stenosis Characteristic facial appearance with short upturned nose, long philtrum, wide mouth, periorbital fullness ‘Cocktail party personality’ (chatty, interactive behaviour) |
| 8q24 | Langer–Giedion | Contiguous gene deletion syndrome characterised by fine sparse hair, brittle nails, bulbous nose and cone-shaped epiphyses on X-ray (owing to deletion of the TRPS1 gene) in addition to exostoses and mental restriction (owing to deletion of neighbouring genes including EXT1) |
| 17p11 | Smith–Magenis | Severe learning difficulties Severe behavioural problems with self-injuring Sleep problems Characteristic facial appearance with square-shaped face and heavy eyebrows |
| 17p13 | Miller–Dieker | Lissencephaly Characteristic facial appearance with vertical furrowing on the forehead, hypertelorism and short nose with anteverted nares |
| 22q11 | DiGeorge/ velocardiofacial/ Shprintzen | Mild to moderate learning difficulties Cardiac anomalies, most commonly tetralogy of Fallot, VSD and interrupted aortic arch Short stature Psychiatric disorders Renal anomalies Cleft palate with or without velopharyngeal insufficiency Hypocalcaemia T cell immune disorder Characteristic facial appearance with tubular nose, narrow palpebral fissures and simple ears |
VSD = ventricular septal defect
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


