Respiratory Physiology and Extracorporeal Life Support
Robert E. Cilley
Division of Pediatric Surgery, Department of Surgery, Pennsylvania State University, Milton S. Hershey Medical Center, Hershey, Pennsylvania 17033.
PULMONARY ANATOMY, PHYSIOLOGY, AND PATHOPHYSIOLOGY
Developmental Anatomy of the Lung
Although all lung disease is not explained by embryology gone awry, an appreciation of the lung in health and disease is based on an understanding of its normal development. The normal function of the lung is dependent on the coordinated development of the airway conducting system in conjunction with a specialized vasculature within an active interstitial matrix. Branching morphogenesis, vasculogenesis, angiogenesis, physical growth, and biochemical maturation must occur in a coordinated fashion. This complex developmental process must be both temporally and spatially controlled.
The uniform nomenclature that is currently used to describe the developmental stages of human lung growth derives from the meeting of the International Congress of Anatomists in Leningrad in 1970. Lung development has been divided into the Embryonic, Pseudoglandular, Canalicular, Saccular, and Alveolar periods based on the histologic appearance (1,2). Key anatomic events in the process of lung development are summarized in Table 11-1 and discussed further in Chapter 61. Molecular regulation of these events is an area of intense active investigation and a brief summary is provided as follows.
Molecular Embryology of Lung Development
The destinies of individual cell lineages that form specific organs have been described in simple organisms. Some of the genes that control these cell lineages have been described. Homologous or similar human genes determine the lung cell-specific lineages that arise from the floor of the hypopharynx in early human lung development. Master regulatory genes control the elaboration of transcription factors that in turn regulate growth factors and receptors that guide lung development. Lung development is environmentally sensitive. Environment–gene interactions are influenced by the structure, shape, and contents of the thorax, as well as the intraluminal pressure of the developing airways and blood flow within the developing vasculature, cellular proliferation, migration, differentiation, maturation, and apoptosis all play a role in this complex process.
Hepatocyte nuclear factors, forkhead factors, thyroid transcription factor-1, sonic hedgehog, certain homeodomain transcription factors, and GATA and Gli transcription factors play significant regulatory roles in lung development. Left–right asymmetry is controlled by transforming growth factor (TGF)-β superfamily members, lefty-1, lefty-2, and Nodal (3). Adhesive glycoproteins, collagen, proteoglycans, and other components of the extracellular matrix, such as matrix metalloproteinases (MMPs) and their inhibitors, also play key roles in the mesenchymal–epithelial–endothelial interactions that are responsible for lung organogenesis (4). Fibronectin, an adhesive glycoprotein that forms high-molecular-weight polymers that interact with embryonic cells during migration, differentiation, and organogenesis, is a key molecular component of the extracellular matrix in lung morphogenesis. Peptide growth factors elaborated by mesenchymal cells control epithelial proliferation, whereas the extracellular matrix controls the patterns of branching and growth. Major growth factor families involved in these processes include TGF, fibroblast growth factor, epidermal growth factor, and platelet-derived growth factor. Thyroid hormone, retinoids, and steroid hormones play regulatory roles in the production of several transcription factors, growth factors, and their receptors. Excess or deficient hormone concentrations are implicated in abnormal lung development.
TABLE 11-1 Lung Development. | |
|---|---|
|
Branching of the developing conducting airway system is dependent on interactions with the mesenchyme such that epithelial cells will not undergo branching when separated from lung mesenchyme. Development of the pulmonary vasculature is also intimately tied to the developing airways and is coordinated by molecular “cross-talk” between the developing vasculature/endothelium and airway/epithelium components of the lung (5,6).
The commitment to specific cell types, such as alveolar type II cells, occurs early in development and is also dependent on epithelial–mesenchymal interactions. Autocrine and paracrine interactions between mesenchymal and epithelial cells control the proliferation of cells and the commitment to cell lines. Extracellular MMPs and their inhibitors play an important role in determining the patterns and shape of the developing lung, as well as the development of the pulmonary vasculature. The events of development are precisely coordinated in a time- and position-specific manner (7,8,9,10,11,12,13). It may someday be possible to precisely determine and regulate the gene switches and transcription factors controlling lung development in order to reverse or correct lung diseases.
Respiratory Physiology
The most important and unique contribution of the lung to the maintenance of homeostasis is as the organ of gas exchange with the environment. Although the nonkeratinized skin of premature infants is theoretically capable of some gas exchange and the highly vascularized intestines may transfer some gases across their large epithelial surfaces, for practical purposes, all respiratory gas exchange occurs across the alveolar-capillary barrier of the pulmonary acinus. Under basal conditions, as little as 2 to 3 mL per kg per minute of oxygen need be transferred from the environment to the circulation, whereas under conditions of maximal exercise, as much as 60 mL per kg per minute may be required. With this enormous reserve, the respiratory transfer of gases rarely limits the uptake and use of oxygen or the removal of carbon dioxide by the body. Under conditions of respiratory failure, gas exchange may be limited in such a way that normal levels of oxygen and carbon dioxide are not maintained. Transfer of oxygen across the alveolar-capillary barrier for delivery to the tissues or removal of carbon dioxide from the blood may be inadequate.
Muscular Activity of Normal Respiration
The coordinated activity of skeletal muscle is required both to maintain the patency of the upper airway and to provide energy for the movement of air into and out of the lungs. Normal aerodigestive function requires that the laryngeal inlet be open during respiration and protected during the initial phase of swallowing. The muscles of ventilation include all the truncal muscles that can contribute to changing the intrapleural pressure or altering the volume of the thorax. The function of some muscles is directed primarily at ventilation (diaphragm, intercostals); other muscles are normally a part of ventilation but have other important functions (abdominal wall muscles); and still other muscles normally provide little assistance with ventilation, except perhaps to stabilize the movement of the thorax, and are called into play under pathologic conditions (accessory muscles of respiration).
The diaphragm is dome shaped and has a longer vertically oriented component than usually is depicted in anatomic diagrams, a fact that is well known to surgeons who perform tube thoracostomy placement or operations in the upper retroperitoneum. The zone of diaphragmatic apposition refers to this long, vertically oriented portion of the diaphragm that lies in direct contact with the inner surface of the thorax. The zone of apposition is disproportionately short in infants compared with adults. Diaphragmatic contraction primarily pulls the diaphragm downward, increasing the intrathoracic volume. The vertical orientation of the zone of apposition provides maximal mechanical advantage during inspiration. Lowering of the intrapleural pressure that occurs with contraction and vertical descent of the diaphragm during inspiration tends to collapse the portion of the rib cage that overlies the lungs. This phenomenon is especially apparent in infants, with their compliant chest walls, and may limit the effectiveness of diaphragmatic function during respiratory failure.
The muscles of the chest wall change the anteroposterior and transverse diameter of the chest by elevating and depressing the ribs, thus increasing or decreasing the
intrathoracic volume. The muscles of inspiration rotate the ribs about their costovertebral articulation such that the sternal end of the rib moves up and the middle of the rib moves upward and outward. (These movements have been called the pump-handle and bucket-handle motion of the ribs.) The ribs are much more horizontally oriented in infants than in older children, so active rib movement contributes less to ventilation. The muscles of inspiration include the levator costae, external intercostals, parasternal intercostals, scalenes, and sternocleidomastoids. The muscles of expiration rotate the ribs downward and inward, and include the subcostals, internal intercostals, transversus thoracis, rectus abdominis, oblique and transverse abdominal muscles, and quadratus lumborum. Muscles of the shoulder girdle can also be recruited for respiration.
intrathoracic volume. The muscles of inspiration rotate the ribs about their costovertebral articulation such that the sternal end of the rib moves up and the middle of the rib moves upward and outward. (These movements have been called the pump-handle and bucket-handle motion of the ribs.) The ribs are much more horizontally oriented in infants than in older children, so active rib movement contributes less to ventilation. The muscles of inspiration include the levator costae, external intercostals, parasternal intercostals, scalenes, and sternocleidomastoids. The muscles of expiration rotate the ribs downward and inward, and include the subcostals, internal intercostals, transversus thoracis, rectus abdominis, oblique and transverse abdominal muscles, and quadratus lumborum. Muscles of the shoulder girdle can also be recruited for respiration.
Pulmonary Mechanics and Respiratory Gas Flow and Distribution
Respiratory gas exchange is dependent on the flow of gases from the environment to the alveolus, where gas exchange takes place. Gas flow is possible only when there is a pressure gradient between the airway and the alveolus. During spontaneous respiration, that pressure gradient is the result of negative intrapleural pressure that results from the muscular activity of the chest wall and the diaphragm. The energy requirements for the movement and distribution of respiratory gases normally constitute less than 3% of the total body energy expenditure. Under conditions of respiratory failure, this energy expenditure can increase many times that amount and may not be sustainable. Respiratory work, the work of breathing, is a form of pressure–volume work. Pressure is required to move a tidal volume of fresh gas into and out of the thorax. The work of spontaneous breathing can be summarized for one breath as:
W = (Pi + Pe)V
where W is work of breathing, Pi is pressure required to move fresh gas into the thorax, and Pe is pressure required to move a tidal volume (V) of fresh gas out of the thorax.
During spontaneous breathing and mechanical ventilation, the expiratory component of the work of breathing usually is zero because exhalation is largely passive as a result of the elastic properties of the lungs and chest wall. It may be significant in obstructive airway disease when active muscular activity is used to aid exhalation. The work of breathing is increased when higher pressures are needed to generate a given tidal volume, minute ventilation increases by an increase in the tidal volume, or minute ventilation increases by higher respiratory rates.
Work, by definition, is the application of force over a distance. The force applied by the respiratory muscles is exerted to overcome two major forces: the total respiratory compliance (contributed by the lung parenchyma, the chest wall, and the abdomen) and the resistance to ventilatory gas flow in the large and small airways. Compliance is defined as the lung volume change that results from a given change in pressure, and it reflects the elastic properties of the entire ventilatory apparatus:
C = ΔV/ΔP
The pressure that must be generated to move a given tidal volume increases as compliance decreases. Any acute parenchymal lung process, such as pneumonia, systemic inflammatory response syndrome, or hydrostatic pulmonary edema, results in a decrease in lung parenchymal compliance. A decrease in compliance is probably the first and most sensitive indicator of lung injury (14). Similarly, abdominal distention, ascites, bony abnormalities of the thorax (e.g., severe kyphoscoliosis), pleural effusions, and generalized edema also may contribute to decreased total respiratory compliance.
In addition to the pressure needed to overcome the compliance of the respiratory apparatus, pressure is also needed to overcome the resistance to flow within the airways. Resistance is defined by the pressure gradient required to produce a given flow within a conducting system:
R = Δ P/flow
Resistance is the net result of factors that tend to retard the flow of gas through the airways and is determined by the physical properties of the respiratory gases (density and viscosity), the length and diameter of the conduit through which they must travel, and the velocity of gas flow. As airway resistance increases, more pressure is required to maintain respiratory gas flow. Airway diameter is the most critical determinant of resistance, and any condition that narrows the airway will have a large effect on resistance. Even under ideal circumstances of laminar flow of respiratory gases within the airways, resistance changes inversely with the fourth power of the diameter of the airway. Therefore, small changes in the airway diameter can have large effects on resistance and on the pressure (i.e., work) required to move tidal volumes. Turbulent flow, which is always present in large airways, accentuates the effect of airway diameter on resistance. Similarly, disease states that result in airway narrowing (bronchospasm) markedly affect the resistive work of breathing. The contribution of the peripheral airways to total airway resistance is proportionally greater in infants and small children than in adults. As a result, they are more susceptible to respiratory failure in diseases that affect the small airways, such as bronchiolitis. In addition, when airway resistance increases, the compliant chest walls of infants may limit their ability to generate adequate pressure to maintain gas flow.
To describe the mechanics of respiration, the lung can be thought of as a compliant reservoir that empties and fills according to first-order kinetics. The time course of volume
change in a compliant reservoir that obeys first-order kinetics is defined by the compliance of the reservoir and the resistance to flow into or out of the reservoir according to the natural logarithmic function:
change in a compliant reservoir that obeys first-order kinetics is defined by the compliance of the reservoir and the resistance to flow into or out of the reservoir according to the natural logarithmic function:
Vτ = V0e±(1/RC)τ
where Vτ is volume at a given time; V0 is initial volume; R is resistance; C is compliance. The sign of the exponent, is positive during filling and negative during emptying; τ is the time constant.
The time constant of such a compliant reservoir is defined as the product of its compliance and resistance. It is a measurement of the time required to equilibrate lung volume and airway pressure after a rapid change in airway pressure, such as occurs at the beginning of inspiration and at the beginning of expiration. According to the equation, one time constant in emptying is the time required to reduce the volume of the lungs to e-1 of their original volume (i.e., to 63% of their original volume). Three time constants are required to equilibrate 95% of a tidal volume and five time constants are required to equilibrate 99% of a tidal volume. Normally, the time constant of the lung is short in comparison with inspiratory and expiratory times, and only becomes important in disease states.
Diseases that decrease compliance, such as respiratory distress syndrome (RDS), result in a shortening of the time constant. The time required to equilibrate the volume of the lung is shortened, inflation and deflation are completed in a shorter time than in normal lungs, and higher respiratory rates may be tolerated than in normal lungs. Likewise, patients with high expiratory resistance, such as occurs in children with reactive airway disease and in infants with bronchiolitis, have a long time constant and rapid respiratory rates may not allow sufficient time for expiration. This phenomenon results in elevation of the lung volume at end expiration [elevated functional residual capacity (FRC)] and may manifest itself as air trapping, carbon dioxide retention, impaired venous return to the heart, and reduced cardiac output.
In an ideal elastic reservoir, the pressure–volume relation is the same whether the reservoir is expanding or contracting. In the lung, the time constant (i.e., the resistance and compliance) that characterizes inflation (inhalation) differs from the time constant of deflation (exhalation). This physical property of the lungs is responsible for the phenomenon of hysteresis. At a given distending pressure, the lung volume is different depending on whether the lung is being inflated from FRC or deflated from maximum volume. Further, the human lung is not an ideal compliant reservoir and does not obey first-order kinetics perfectly. In both health and disease, the lung behaves like a reservoir with multiple compartments in parallel, each of which has a different time constant. The behavior of the lung as a whole is a summation of the multiple compartments.
When the lung is inflated with positive pressure from FRC, collapsed alveoli or atelectatic areas require the application of relatively high pressures to open (15,16). Initial compliance is low, corresponding to the initially flat portion of the compliance curve seen in Fig. 11-1, and is referred to as starting compliance (Cstart). The portion of the lung that expands in this fashion is referred to as recruitable and represents areas that may be maintained open under the influence of positive end-expiratory pressure (PEEP) when mechanical ventilation is required. The normal lung has few areas of atelectasis and, therefore, little in the way of recruitable volume. However, when subjected to positive-pressure ventilation, even normal lungs develop atelectasis if they are allowed to equilibrate to atmospheric pressure at end expiration, such as occurs when endotracheal intubation is used without PEEP. Diseased lungs may have significant amounts of recruitable volume. The next portion of the compliance curve is steep, representing the distention of normal alveoli, which requires only small changes in pressure to effect relatively large increases in volume. This is where normal respiration takes place. When all recruitable alveoli have been fully distended, large pressures are required to effect a small change in volume. The compliance curve again becomes flat above this second inflection point. In normal lungs, this is the total lung capacity. Even diseased lungs in patients with “diffuse” lung injury, such as adult RDS, have small portions that retain near-normal compliance characteristics. This is the portion of the lung that is preferentially
ventilated and also the portion that is most vulnerable to injury from high-pressure mechanical ventilation. When high pressures are used in conjunction with mechanical ventilation, the more normal portion of the lung is most likely to be overdistended and injured.
ventilated and also the portion that is most vulnerable to injury from high-pressure mechanical ventilation. When high pressures are used in conjunction with mechanical ventilation, the more normal portion of the lung is most likely to be overdistended and injured.
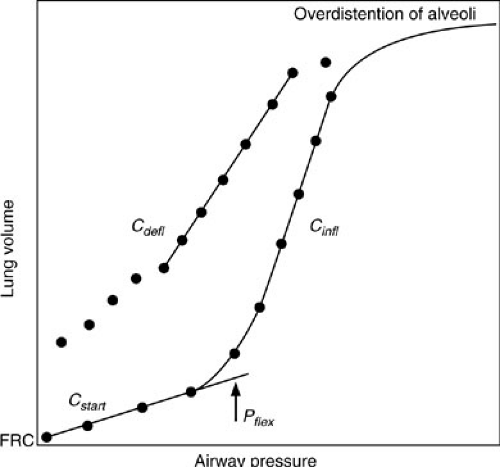 FIGURE 11-1. Static pressure-volume curve. Cstart reflects the poor distensibility of the lung at low volumes. High pressures are needed to open airways that are closed. Pflex is the point at which recruitable alveoli are open. Cinfl is the compliance of the respiratory system when all recruitable airspaces are open. Compliance decreases dramatically when the lung is overdistended. In injured lungs, overdistention may occur at relatively small total volumes. Cdefl differs from the inflation limb because of hysteresis. FRC, functional residual capacity. |
Pulmonary Blood Flow and Pulmonary Vascular Resistance
During intrauterine life, only about 10% of the right ventricular output actually circulates through the pulmonary vasculature. Most of the blood flow is directed to the systemic circulation at the level of the ductus arteriosus and foramen ovale. In spite of this lack of blood flow, the pulmonary vasculature develops with the potential to receive all the cardiac output almost immediately after birth, when the transition to pulmonary gas exchange must occur abruptly. This is a truly remarkable developmental design in which form anticipates function.
Pulmonary blood flow increases with the first expansion of the lungs that occurs with the first breath. Pulmonary vascular resistance falls dramatically and the ductus arteriosus closes over the course of several days. Less blood also is shunted at the atrial level and the infant achieves true parallel circulation between the pulmonary and systemic circulations. “Fetal” circulation persists (persistent pulmonary hypertension of the newborn) when pulmonary vascular resistance remains elevated and blood is shunted at the atrial, ductal, and intrapulmonary levels, resulting in admixture of venous blood to the systemic circulation and hypoxia. Because hypoxia is one of the most potent stimuli of pulmonary vasoconstriction, a deteriorating cycle may begin. Acidosis and hypercarbia that accompany the hypoxia also may contribute to the pulmonary vasospasm. Excessive muscularization of the pulmonary arteries that is characteristic of the lungs in congenital diaphragmatic hernia contributes to pulmonary hypertension. Pulmonary hypoplasia, with a smaller cross-sectional area of pulmonary vasculature, may also contribute to pulmonary hypertension.
The pulmonary vasculature is highly compliant and can accept large changes in blood flow with minimal changes in pressure. Pulmonary blood flow is highly dependent on gravity. The effect of gravity is to create a hydrostatic gradient that favors blood flow to the dependent regions of the lung (17). The zones of the lung describe the relations among pulmonary arterial pressure, alveolar pressure, and pulmonary venous pressure. In the upper areas of the lung (zone 1), alveolar pressure theoretically exceeds arterial pressure, and there is no flow. It is unlikely that this occurs in healthy lungs, but it may be important during mechanical ventilation that exposes the alveoli to extremely high pressures. In zone 2, the gradient between arterial pressure and alveolar pressure determines pulmonary blood flow; both are greater than pulmonary venous pressure. Most of the lung operates under zone 3 conditions in healthy individuals. Here, flow is determined by the pulmonary arteriovenous pressure gradient; both pressures exceed alveolar pressure.
Gas Exchange: The Functions of Oxygenation and Ventilation
Under normal conditions, oxygenation and ventilation occur simultaneously and harmoniously. Apnea results in both hypoxia and hypercarbia, while restoration of breathing reestablishes gas exchange. This phenomenon is so much a part of everyday experience that it is intuitive to think of oxygenation and ventilation together. However, oxygenation and ventilation are separable functions of the lung, and under pathologic conditions, may even interfere with each other (18).
Oxygen and carbon dioxide are transferred across the alveolar-capillary membrane by passive processes of diffusion. Gas transfer is proportional to the difference between the partial pressures of the gas in the alveolus and the pulmonary capillary blood, and is similar for oxygen and carbon dioxide. Only about one-third of the 0.75 sec that the blood spends in the pulmonary capillary are necessary for equilibration; diffusing capacity must be reduced significantly to result in abnormal pulmonary gas exchange. Abnormal thickening of the alveolar-capillary membrane, reduced transit time in the capillary, and alveolar hypoxia may contribute to inadequate gas exchange. Increases in the fraction of inspired oxygen (FIO2) overcome hypoxemia resulting from diffusion abnormalities.
Carotid body and central nervous system (CNS) chemoreceptors continuously monitor the arterial partial pressure of carbon dioxide (PaCO2) and adjust alveolar ventilation to maintain a value within a narrow range, regardless of the amount of metabolic carbon dioxide produced. Hyperventilation is defined as excessive alveolar ventilation for the level of carbon dioxide production. Hypoventilation results when alveolar ventilation is inadequate to remove metabolically produced carbon dioxide and hypercarbia results. An increase in dead-space ventilation (reducing alveolar ventilation for the same minute ventilation) or lack of normal coupling between the PaCO2 and minute ventilation by the respiratory centers of the brain (e.g., CNS depression, brainstem injury, neuromuscular disease, musculoskeletal disease of the chest, upper airway obstruction) may result in hypoventilation. Any elevation of the alveolar partial pressure of carbon dioxide (PCO2) obligatorily causes a fall in the alveolar partial pressure of oxygen (PO2), as defined by the alveolar gas equation (simplified form):
PalveolarO2 = PinspiredO2 – (Palveolar CO2/respiratory quotient)
The relation between ventilation and perfusion (VA/Q) determines to a large degree the adequacy of pulmonary gas exchange. The relation may vary from areas of the lung with no ventilation and some blood flow (VA/Q = 0, true
shunt) to areas with ventilation and no perfusion (VA/Q = infinity, dead space). Normally, the mean VA/Q is 1, varying from 0.6 to 3. The most common VA/Q abnormality is a reduction. The respiratory center responds by increasing minute ventilation, which is preferentially distributed to already ventilated areas, resulting in a fall in their postcapillary PaCO2 and a rise in their postcapillary PO2. This does not effect a rise in the arterial partial pressure of oxygen (PaO2) because the arterial oxygen content (CaO2) of the postcapillary blood is changed little by a rise in the PO2 as most of the oxygen is carried by the already saturated hemoglobin. However, the PaCO2 is normalized because carbon dioxide content is proportional to the PCO2. If the VA/Q abnormality becomes too great, minute ventilation cannot compensate and PaCO2 will rise. If the rise in the PaCO2 is gradual, it may be well tolerated and part of the compensation for respiratory failure because it increases the efficiency of the compromised respiratory system (the higher the alveolar PCO2, the more CO2 eliminated at a given minute ventilation). Marked elevations in PaCO2 can be tolerated if they occur gradually and are compensated. If the rise in the PaCO2 is acute and results in CNS depression and acidosis, alveolar ventilation must be assisted. The hypoxemia of VA/Q abnormalities responds to supplemental oxygen administration, becoming more resistant as the abnormality worsens.
shunt) to areas with ventilation and no perfusion (VA/Q = infinity, dead space). Normally, the mean VA/Q is 1, varying from 0.6 to 3. The most common VA/Q abnormality is a reduction. The respiratory center responds by increasing minute ventilation, which is preferentially distributed to already ventilated areas, resulting in a fall in their postcapillary PaCO2 and a rise in their postcapillary PO2. This does not effect a rise in the arterial partial pressure of oxygen (PaO2) because the arterial oxygen content (CaO2) of the postcapillary blood is changed little by a rise in the PO2 as most of the oxygen is carried by the already saturated hemoglobin. However, the PaCO2 is normalized because carbon dioxide content is proportional to the PCO2. If the VA/Q abnormality becomes too great, minute ventilation cannot compensate and PaCO2 will rise. If the rise in the PaCO2 is gradual, it may be well tolerated and part of the compensation for respiratory failure because it increases the efficiency of the compromised respiratory system (the higher the alveolar PCO2, the more CO2 eliminated at a given minute ventilation). Marked elevations in PaCO2 can be tolerated if they occur gradually and are compensated. If the rise in the PaCO2 is acute and results in CNS depression and acidosis, alveolar ventilation must be assisted. The hypoxemia of VA/Q abnormalities responds to supplemental oxygen administration, becoming more resistant as the abnormality worsens.
The admixture of venous blood with blood returning from ventilated alveolar-capillary units results in a shunt. The normal “physiologic” shunt is about 1% to 3% of the cardiac output and is a result of the return of the bronchial circulation to the left side of the heart and to the intracardiac veins (thebesian veins) that return to the left ventricle. High pressure in the pulmonary arterial circulation predisposes to shunting. Blood that passes through the capillaries of collapsed alveoli (atelectasis) or those filled with inflammatory exudate or edema fluid results in a shunt. Pulmonary arteriovenous malformations and cyanotic congenital heart disease also result in shunts. The hypoxemia associated with shunted blood is resistant to increases in the FIO2. Even breathing 100% oxygen makes only a small change in the PaO2 in the presence of a true shunt.
Under normal circumstances, a fall in the mixed venous PO2 such as that which occurs during exercise or other states of increased oxygen consumption (VO2) does not result in hypoxia. Ventilation is normally able to increase far greater than cardiac output. In lung disease, gas exchange may not be able to improve during exercise or stress to maintain oxygenation of the blood (i.e., VA/Q falls) and hypoxia results.
Pulmonary Surfactant
It was recognized in the 1950s that the lungs of premature infants with respiratory distress were deficient in substances called pulmonary surfactant. Pulmonary surfactant, by reducing surface tension at the air–liquid interface of the alveolar lining, stabilizes the alveoli at low lung volumes and prevents alveolar collapse at end expiration. RDS of the newborn is characterized by surfactant deficiency and alveolar collapse (atelectasis). It occurs primarily in premature infants, but others, such as infants of diabetic mothers, also are at risk. RDS is the leading cause of perinatal mortality and morbidity in developed countries. Its high prevalence, treatment, and prevention have enormous economic and social implications.
All mammalian species display augmented surfactant production after about 85% of gestation is complete. Pulmonary surfactant is produced in the type II alveolar lining cells that comprise 10% to 15% of the cell population of the lung. The surfactant-producing type II cells are distinguished ultrastructurally by the presence of lamellar bodies, which are the intracellular storage form of surfactant lipids. Lamellar bodies are secreted into the alveolar space by the process of exocytosis. Conformational changes then occur in the lamellar body substances that facilitate the spreading of surfactant over the alveolar surface.
Pulmonary surfactant has a lipid component and a protein component. The lipid component accounts for 90% of the mixture by weight and is primarily composed of glycerophospholipids, of which dipalmitoylphosphatidylcholine is the most abundant compound and the major surface-active component. Surfactant contains several proteins that assist in its distribution over the alveolar surface and contribute to its surface-active properties (19).
The major protein component of pulmonary surfactant is a 28,000- to 38,000-molecular-weight glycoprotein (surfactant protein-A, SP-A) that is highly conserved across mammalian species. SP-A has the ability to bind lipids and carbohydrates, and to bind to specific cell surface receptors. SP-A, along with other surfactant proteins (SP-B, SP-C, SP-D), promotes the rapid formation of surfactant surface films that coat the alveolus and reduce surface tension. SP-A is also important in the binding and recycling of surfactant material by the type II cells through the process of endocytosis that involves specific receptors for SP-A.
The synthesis of the lipid component of pulmonary surfactant is subject to multifactorial control, and many substances, including glucocorticoids, prolactin, thyroid hormones, estrogens, androgens, insulin, and catecholamines acting through cyclic adenosine monophosphate, are important in its control (20). The genes encoding SP-A, SP-B, and SP-C are regulated independently during fetal lung development. SP-A gene expression is undetectable in the human lung before 20 weeks of gestation. Differentiated type II cells that contain a few lamellar bodies are observed by 22 weeks, and active surfactant secretion begins to occur after 30 weeks of gestation. The expression of SP-B and SP-C is detectable much earlier in development than that of SP-A. Cyclic adenosine monophosphate and glucocorticoids play an important role in the regulation of SP-A gene expression.
Term infants of diabetic mothers have an increased incidence of RDS. Fetal hyperinsulinemia in response to maternal hyperglycemia may play a role in the abnormal development of surfactant-producing capability. It also has been observed that fetal “stress” associated with maternal hypertension and uteroplacental insufficiency results in accelerated lung maturation. Such infants usually are small and yet have a decreased incidence of RDS. Elevated levels of glucocorticoids and other stress hormones, such as catecholamines, enhance lung maturation and accelerate SP-A expression, phospholipid synthesis, and surfactant secretion. Antenatal glucocorticoid treatment results in enhanced maturation of the lung and is used therapeutically to promote lung maturation when preterm delivery is anticipated.
In addition to surface-active properties, surfactant proteins are involved in the processing (excretion, uptake, metabolism) of surfactant and also have immunologic functions. Deficiencies in surfactant proteins and genetic heterogeneity alter lung function and the response to infection and lung injury. SP-A has both surface-active and immunity functions. SP-B is primarily involved in surfactant lipid processing. SP-C is involved in surfactant metabolism and processing, whereas SP-D functions pertain primarily to innate immunity (21). Genetic variations may alter the risk of RDS and affect the host response to inflammatory lung injury and infection.
Non–gas-exchange Functions of the Lung
The lung is more than simply a gas-exchanging organ. It is composed of living cells and accounts for 1% to 3% of the body’s VO2. The lung is ideally suited as a metabolic organ because the entire cardiac output must contact the pulmonary vascular endothelium. Substances metabolized or altered by the pulmonary vascular endothelium include angiotensin I, bradykinin, prostaglandins, serotonin, and norepinephrine (22).
The lung also serves as a filter, removing particulate debris, thrombi, and bacteria from the circulation. The lung is a major determinant of right ventricular afterload and left ventricular preload. It provides buoyancy in aquatic environments.
Respiratory Pathophysiology
Respiratory failure in infants and children is caused by many diseases, and a complete discussion is beyond the scope of this chapter. Several pathologic processes and diseases are discussed as follows because of their particular relevance to pediatric surgeons.
Abnormalities of Lung Development
Developmental abnormalities can occur in relation to any of the components of the lung. These are summarized here and discussed in detail elsewhere in the text. Abnormal development of the large airways may result in atresia or agenesis of the trachea or bronchi, which may result in respiratory failure at birth. In pulmonary sequestration, lung tissue develops separate from the tracheobronchial tree and in association with an abnormal blood supply from the systemic circulation. Bronchogenic cysts contain respiratory epithelium lining a fibromuscular wall that may contain cartilage and muscle. They may communicate with a bronchus or be entirely separate. Cystic adenomatoid malformations are characterized by disorganized parenchymal lung growth that includes a marked increase in tissue at the terminal bronchiolar level, lined by abnormal ciliated columnar epithelium, surrounded by an interstitium with disorganized elastic tissue and smooth muscle, and without mature alveoli. Vascular abnormalities such as arteriovenous malformations are treated with surgery or embolization. Disordered lymphatic development (pulmonary lymphangiectasia) may result in chylothorax and respiratory failure.
Lung Hypoplasia
A variety of newborn anomalies of great interest to pediatric surgeons are associated with lung hypoplasia. The study of hypoplastic lung development may hold the key to therapeutic strategies that may be helpful in treating many different diseases. Hypoplastic lung development, in which the lung is inadequately developed relative to the gestational age, is usually found in association with other fetal anomalies, but can occur in isolation. There is a spectrum of hypoplastic lung development ranging from severe forms that are incompatible with survival to mild forms in which the diagnosis is only suspected. Hypoplastic lung development is distinguished from the respiratory insufficiency of prematurity because the latter represents normal lung development interrupted by the premature institution of pulmonary gas exchange; however, the parallels are obvious and therapies may be similar.
The pathologic criteria for establishing the diagnosis of pulmonary hypoplasia include a lung-to-body weight ratio more than one standard deviation below normal (normal, 0.018 ± 0.003) and a low morphometric radial alveolar count (number of alveoli encountered on a straight line drawn between a bronchiole and the periphery of the acinus; low-normal, 4.1). These measurements are not clinically helpful. Prenatal estimations of pulmonary hypoplasia have been made using ultrasound to measure the fetal chest circumference-to-abdominal circumference ratio, with good correlation to postnatal pathologic findings (23). The modulation of fetal ductus arteriosus flow by fetal breathing movements has also been proposed as a clinically useful predictor of pulmonary hypoplasia (24). The fetal lung-to-head ratio may predict poor outcome in congenital diaphragmatic hernia (25).
TABLE 11-2 Conditions Associated with Pulmonary Hypoplasia. | ||
|---|---|---|
|
Pulmonary hypoplasia occurs in association with many other conditions (Table 11-2) (26). Prolonged compression of the fetal thorax has been postulated to explain pulmonary hypoplasia in oligohydramnios associated with renal agenesis (Potter syndrome), urinary obstruction, and cases of prolonged amniotic fluid leak. Intrathoracic compression caused by fluid or a mass effect similarly could explain pulmonary hypoplasia associated with a space-occupying lesion of the chest. Absent or reduced fetal breathing movements have also been associated with pulmonary hypoplasia. Agenesis of the phrenic nerve and agenesis of the muscle within the diaphragm illustrate this most clearly. It is also possible that amniotic fluid contains growth factors that are necessary for normal lung development.
Experimental models have duplicated some of these clinical observations, including the surgical creation of diaphragmatic hernia in fetal lambs and the teratogenic creation of diaphragm defects with nitrofen (27,28). Fetal lamb tracheostomy resulting in unrestricted egress of fetal lung fluid causes pulmonary hypoplasia, whereas fetal pharyngostomy does not, implying that distending pressure in the developing fetal airways is of critical importance to normal lung development (29,30). Further evidence of the importance of “back pressure” in the developing airways comes from the observation that tracheal ligation may reverse the hypoplastic development of experimentally created diaphragmatic hernia (31). Fraser syndrome (simultaneous occurrence of renal agenesis and laryngeal atresia), a fatal anomaly, results in nonhypoplastic, more normal lung development (32).
Treatment strategies designed to prevent pulmonary hypoplasia include in utero shunting of intrathoracic fluid, intraamniotic fluid instillation to counteract the effects of oligohydramnios, fetal surgery to remove space-occupying lesions of the thorax, fetal diaphragmatic hernia repair, temporary tracheal ligation or balloon occlusion to promote lung growth, and fetal urinary tract diversion. The therapeutic implications of manipulating the “airway pressure” of the fetus and the newborn as a method of promoting lung development have been studied both experimentally and clinically (25).
Respiratory Failure of Prematurity
The respiratory failure associated with premature birth is an enormous health problem, accounting for most admissions to neonatal intensive care units (ICUs) in developed countries. The primary pathology is surfactant deficiency resulting in elevated alveolar surface tension, alveolar collapse, atelectasis, and respiratory insufficiency. The use of mechanical ventilation to support gas exchange may result in further lung injury. The greatest therapeutic advance in the care of RDS of prematurity has been the use of exogenous surfactant. Its use has resulted in a measurable
decrease in premature infant mortality (33,34). Premature infants treated for respiratory failure with supplemental oxygen, endotracheal intubation, and mechanical ventilation are at risk for the development of bronchopulmonary dysplasia. Bronchopulmonary dysplasia is defined clinically as oxygen dependence that is present for more than 28 days after the use of mechanical ventilation, along with persistent abnormalities on the chest radiograph. Pathologic findings include epithelial necrosis, squamous metaplasia, organization of hyaline membranes in the airways, and fibroblast proliferation in the lung interstitium. The beginnings of these changes may be seen as soon as a few days after birth. The relative contribution of oxygen, airway pressure, and intubation to the pathogenesis are unknown. In its worse form, chronic lung disease results in pulmonary hypertension, cor pulmonale, and death. This severe form is relatively uncommon; however, less severe forms that result in chronic oxygen dependence or the need for prolonged mechanical ventilation with ultimate recovery are common.
decrease in premature infant mortality (33,34). Premature infants treated for respiratory failure with supplemental oxygen, endotracheal intubation, and mechanical ventilation are at risk for the development of bronchopulmonary dysplasia. Bronchopulmonary dysplasia is defined clinically as oxygen dependence that is present for more than 28 days after the use of mechanical ventilation, along with persistent abnormalities on the chest radiograph. Pathologic findings include epithelial necrosis, squamous metaplasia, organization of hyaline membranes in the airways, and fibroblast proliferation in the lung interstitium. The beginnings of these changes may be seen as soon as a few days after birth. The relative contribution of oxygen, airway pressure, and intubation to the pathogenesis are unknown. In its worse form, chronic lung disease results in pulmonary hypertension, cor pulmonale, and death. This severe form is relatively uncommon; however, less severe forms that result in chronic oxygen dependence or the need for prolonged mechanical ventilation with ultimate recovery are common.
The best therapy for bronchopulmonary dysplasia is prevention, through reduction in premature birth and avoidance of excessive oxygen exposure, along with strategies of mechanical ventilation that reduce lung injury. Once bronchopulmonary dysplasia is established, these infants require an approach to care that is somewhat different than the acute care phase of the respiratory failure of prematurity. Weaning strategies from mechanical ventilation include such concepts as “weaning schedules,” in which ventilator changes are made according to a predesigned plan. Continuous monitoring using pulse oximetry, along with the ability of the bedside caregiver (nurse or parent) to make immediate changes in the FIO2 in response to changes in oxygen saturation, prevents prolonged periods of hypoxia and hyperoxia. Nutritional support with both parenteral and enteral nutrition to ensure progressive accrual of lean body mass is of paramount importance. Pharmacologic therapy with antibiotics and bronchodilators is used as needed. Infant stimulation is an essential part of therapy for these infants, who are at increased risk for developmental delay. The use of systemic corticosteroids may result in improvement in lung function and facilitate weaning from mechanical ventilation; however, repeated and high-dose steroid use in infants may have long-term side effects on brain and lung development (35,36,37). The effects of steroid use on somatic growth are variable (37). The role of tracheostomy remains uncertain. Any infant who will require mechanical ventilation for more than 1 month should be considered for tracheostomy.
Acute Lung Injury
The lung, in injury and disease, assumes a role of critical importance perhaps more than any other organ. The lung may be developmentally abnormal (e.g., hypoplastic) or simply too immature to carry out the function of gas exchange (e.g., respiratory failure of prematurity). The lung may lack normal defense mechanisms as a result of genetic abnormalities such as cystic fibrosis or immune deficiencies. The lung may be primarily injured as a result of infection, aspiration of gastric contents, or externally applied forces (e.g., blunt injury from trauma). The lung may function abnormally as a result of pathologic conditions that target other organ systems and then generate a systemic inflammatory response that results in lung injury (e.g., burns, pancreatitis, tissue necrosis, abscesses, circulating endotoxin or bacteria, ischemia-reperfusion injury). When respiratory failure progresses to the point that gas exchange cannot be maintained, mechanical ventilation and supplemental oxygen are the mainstays of therapy. The treatment of respiratory insufficiency may be injurious to the lung, resulting in ventilator-induced lung injury and oxygen toxicity.
Although there are many types of lung injury, the lung has a limited repertoire of response to injury (38). The general pattern of lung injury is as follows: endothelial permeability increases, fluid and protein accumulate in the interstitium of the lung, and finally alveolar fluid accumulates as a result of increased epithelial permeability. The lung may recover if injury is reversed, or gas exchange may become impaired sufficiently to preclude survival. Under some circumstances, ongoing inflammation leads to fibrosis, resulting in chronic lung disease or late death. Many endogenous mediators and the cells that produce them have been implicated in acute lung injury. The host-defense mechanisms that are protective against some injuries can be “overexpressed” and cause widespread organ injury.
Acute Lung Injury at the Cellular Level
Neutrophils are the most important cell in the development of acute lung injury (39). Neutrophils and their products are present in increased numbers in the bronchoalveolar lavage fluid of patients with acute lung injury. Pathologic specimens show increased numbers of sequestered neutrophils in acute lung injury. Neutrophil depletion attenuates experimental mediator-induced lung injury, as does functional manipulation of neutrophils, such as inhibition of cyclooxygenase. RDS may occur in neutropenic patients, but lung function may worsen when neutropenia resolves. Normally, there is a large pool of neutrophils in the vascular space of the lung. This population of neutrophils increases dramatically under the influence of chemoattractants that are elaborated by endothelial cells, monocytes, and other neutrophils in the injured lung. Neutrophils in the intravascular space contact the endothelial lining cells during their traverse through the pulmonary circulation. Neutrophil adherence and migration are regulated largely by the complementary action of the neutrophil cell surface adhesion molecule
receptors and the vascular endothelial adhesion molecules. Some of these adhesion molecules are expressed constitutively and others are upregulated in response to cytokines [particularly interleukin-1 (IL-1) and tumor necrosis factor (TNF)] and proinflammatory stimuli (see Chapter 14). Neutrophils that accumulate in the lung have increased nuclear accumulation of the transcription factor, nuclear factor-kappa B, that is associated with the expression of a number of proinflammatory cytokines (39). Components of signaling pathways associated with inflammation are upregulated.
receptors and the vascular endothelial adhesion molecules. Some of these adhesion molecules are expressed constitutively and others are upregulated in response to cytokines [particularly interleukin-1 (IL-1) and tumor necrosis factor (TNF)] and proinflammatory stimuli (see Chapter 14). Neutrophils that accumulate in the lung have increased nuclear accumulation of the transcription factor, nuclear factor-kappa B, that is associated with the expression of a number of proinflammatory cytokines (39). Components of signaling pathways associated with inflammation are upregulated.
Neutrophils are larger than pulmonary capillaries and must deform to pass through. Because neutrophils are less deformable when activated, their contact with the pulmonary capillary endothelial cells is enhanced. In acute lung injury, the adherent neutrophils and those recruited into the interstitium of the lung are a major source of oxygen radicals (molecular oxygen, superoxide, hydroxyl) and proteases that result in tissue injury. Neutrophils that leave the vascular space to enter the interstitium may enter the alveolus.
Pulmonary vascular endothelium is also actively involved in acute lung injury. The cell surface adhesion molecules are upregulated in lung injury and affect neutrophil adherence. The contact between neutrophils and endothelial cells is increased and prolonged, providing a microenvironment protected from circulating antioxidants and antiproteases, and favoring endothelial cell injury. With damage to the integrity of the endothelial cell–endothelial cell barrier, permeability increases and interstitial edema results. Impairment of the normal metabolism of serotonin, norepinephrine, prostaglandins, bradykinin, and angiotensin I by the endothelial cells also occurs in lung injury. In addition, endothelial cells produce chemoattractants that augment and perpetuate injury.
The macrophage/monocyte cell line also plays a role in acute lung injury. Resident macrophages respond to inflammatory mediators and become activated. Activated macrophages produce the same toxic products as neutrophils, including oxygen radicals and proteases. Lung macrophages also release TNF and IL-1. The population of interstitial macrophages increases in the first several days after an inflammatory response is initiated. This may be the most important event in the perpetuation of the inflammatory response that results in ongoing lung injury even after the systemic factors that initiated the response have been controlled. The long life of lung macrophages, measured in days, compared with neutrophils, which may only survive for hours, also contributes to the perpetuation of the inflammatory response. Macrophages also play an important role as bactericidal cells and phagocytic cells in injured lungs. Macrophages participate in the regulation of fibroblast function and probably play a role in the fibrosis that results when lung injury is perpetuated.
Platelets have a less certain role in acute lung injury and may be involved secondarily; however, sequestered platelets are commonly found in acute lung injury. Platelet release of serotonin, proteases, and prostaglandins may contribute to lung injury.
Biochemical Mediators of Lung Injury
The cellular components that characterize acute lung injury elaborate products that perpetuate and alter the injury. When neutrophils and macrophages are activated by the complement cascade or other injury, cytokines, phospholipid metabolites, and oxygen radicals are produced. These products influence the activity and function of the cells around them and participate in the activation of enzymatic cascades related to inflammation (see Chapter 14). Among them, cytokines and products of arachidonic acid metabolism are especially important. IL-1, IL-2, IL-6, TNF, and interferon (IFN)-γ are the most important cytokines. The activity of phospholipases is increased in activated inflammatory cells, which in turn increases the cleavage of arachidonic acid from membrane phospholipids. The action of cyclooxygenase on arachidonic acid results in the production of prostaglandins, thromboxanes, and prostacyclin. Thromboxane is a potent agonist for platelet aggregation and smooth muscle contraction, and is implicated in the pulmonary hypertension and bronchoconstriction associated with sepsis and acute lung injury. Prostacyclin functions in an opposing fashion, by inhibiting platelet aggregation and causing vasodilation, and may be in part responsible for the systemic vasodilation associated with the systemic inflammatory response. Other prostaglandins are primarily vasodilators. During lung injury, these compounds and others may have exaggerated local and systemic effects.
Activation of procoagulants also occurs as part of the inflammatory response. Procoagulants are present in blood, lung interstitium, and inflammatory exudates within the airspaces (40). Factor Xa, thrombin, and fibrin interact with inflammatory mediators and influence the tissue response to injury. Therapy directed at procoagulant activity may have therapeutic potential. Surfactant lipid and surfactant protein function and production may also be altered in acute lung injury. Genetic heterogeneity in the control and production of inflammatory mediators in both animal and human populations may explain clinical variations of disease as phenotypic patterns of the inflammatory response. The key events of acute lung injury are summarized in Fig. 11-2.
Overdistention Lung Injury/Pressure-Volume Lung Injury
The use of mechanical ventilation plays a role in lung injury. Patients with normal lungs can be sustained almost
indefinitely using both positive- and negative-pressure mechanical ventilation with minimal deleterious effects. Likewise, long-term animal experiments failed to show pathologic lung changes that can be attributed to mechanical ventilation itself when normal ventilating pressures and volumes were used. In acute respiratory failure, however, mechanical ventilation is being applied to already injured lungs, and abnormally high pressures and tidal volumes may be necessary to achieve adequate gas exchange. Although necessary as a life-sustaining measure to maintain pulmonary gas exchange, the use of mechanical ventilation may be responsible for lung injury. Several lines of evidence, both clinical and experimental, implicate the use of mechanical ventilation as both a culprit and a cure for respiratory failure (41).
indefinitely using both positive- and negative-pressure mechanical ventilation with minimal deleterious effects. Likewise, long-term animal experiments failed to show pathologic lung changes that can be attributed to mechanical ventilation itself when normal ventilating pressures and volumes were used. In acute respiratory failure, however, mechanical ventilation is being applied to already injured lungs, and abnormally high pressures and tidal volumes may be necessary to achieve adequate gas exchange. Although necessary as a life-sustaining measure to maintain pulmonary gas exchange, the use of mechanical ventilation may be responsible for lung injury. Several lines of evidence, both clinical and experimental, implicate the use of mechanical ventilation as both a culprit and a cure for respiratory failure (41).
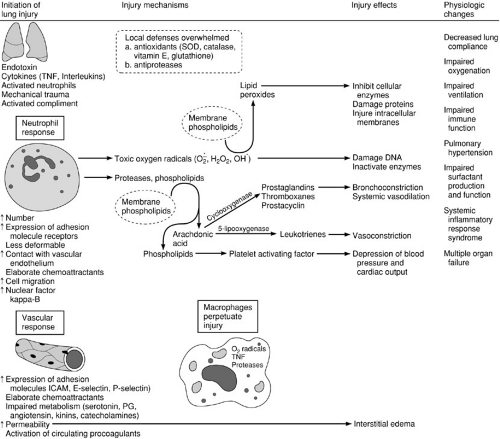 FIGURE 11-2. Summary of the events of acute lung injury. (See text for details.) TNF, tumor necrosis factor; SOD, superoxide dismutase; ICAM, intercellular adhesion molecule; PG, Inflammatory cytokines initiate a neutrophil response that overwhelms local defenses, damages cells, and alters respiratory mechanics. |
The multiinstitutional, randomized, prospective trial of extracorporeal membrane oxygenation (ECMO) for adult respiratory failure sponsored by the National Institutes of Health concluded that ECMO was not more effective in the treatment of severe respiratory failure than conventional mechanical ventilation (42). A more appropriate conclusion would have been that when life is sustained using ECMO and irreversible lung injury has already taken place, survival is not improved. ECMO was applied more
appropriately to neonates, for whom the methods of mechanical ventilation available at the time were failing. In this population, respiratory failure that was rapidly fatal using conventional means was treated with extracorporeal support and “lung rest.” The rapidity with which respiratory failure progressed to become life threatening may be the feature that allowed “lung rest” to result in recovery, before the effects of high pressures and high oxygen concentrations became irreversible. During ECMO, the mechanical ventilator was turned down to pressures that presumably were not injurious. The concept that “lung rest” could result in the complete reversal of fatal respiratory failure of short duration emphasized that mechanical ventilation itself may be injurious to the lung. In neonates with severe respiratory failure, simply eliminating mechanical ventilation can result in the rapid return of respiratory function within a few days.
appropriately to neonates, for whom the methods of mechanical ventilation available at the time were failing. In this population, respiratory failure that was rapidly fatal using conventional means was treated with extracorporeal support and “lung rest.” The rapidity with which respiratory failure progressed to become life threatening may be the feature that allowed “lung rest” to result in recovery, before the effects of high pressures and high oxygen concentrations became irreversible. During ECMO, the mechanical ventilator was turned down to pressures that presumably were not injurious. The concept that “lung rest” could result in the complete reversal of fatal respiratory failure of short duration emphasized that mechanical ventilation itself may be injurious to the lung. In neonates with severe respiratory failure, simply eliminating mechanical ventilation can result in the rapid return of respiratory function within a few days.
When the use of routine mechanical ventilation was still in its infancy, Greenfield and colleagues demonstrated that pulmonary edema developed in dogs that were subjected to high-volume mechanical ventilation using a pressure of 30 cm H2O for several hours (43). This was the first study that demonstrated that mechanical ventilation could alter lung function and cause lung injury. The morphologic characteristics of the lung injury associated with the application of high pressure to normal lungs in rats included injury that progressed from perivascular edema, to interstitial edema with detachment of endothelial cells from their basement membrane, to type I cell damage, denuding of the epithelial side of the basement membrane, alveolar edema, and the formation of hyaline membranes consisting of cellular debris and protein (44). The question of whether pressure or volume of distention was the primary culprit responsible for this kind of lung injury then was addressed. Using microvascular permeability changes to assess lung injury, positive- and negative-pressure ventilation (using a small “iron lung”) to equivalent degrees of lung overdistention (40 to 45 mL per kg) resulted in identical lung injuries (45). The investigators further showed that thoracoabdominal strapping ablated the injury associated with high-pressure ventilation. Others have demonstrated in similar experiments that when lung overexpansion is prevented (using a body cast), high airway pressures are tolerated without lung injury, and that when low pressures are applied to the lung and there is no restriction to its expansion (ex vivo), severe alterations in microvascular permeability result (46). Dreyfuss and Saumon also demonstrated that the most important determinant of overdistention lung injury is the absolute lung volume at end inspiration, with similar injuries produced by high-FRC/low-tidal volume ventilation and low-FRC/high-tidal volume ventilation (47). Large swings in tidal volume with zero end-expiratory pressure are not tolerated as well, and PEEP tends to preserve gas exchange and reduce edema to some extent. PEEP decreases the shear stress associated with the repetitive opening and closing of the terminal airways. Overdistention can cause injury beyond the lung itself, and Kolobow and colleagues demonstrated that prolonged mechanical ventilation with excessive tidal volumes resulted in a syndrome of multiple organ failure (48).
It is clear from these experiments that the most significant determinant of lung injury in normal lungs exposed to excessive pressures and volumes is the degree of alveolar distension present at end inspiration. The mechanism by which this injury takes place is not clear. However, striking ultrastructural similarities have been shown between the endothelial and epithelial lesions that result from elevated capillary transmural pressure and the lesions of lung airway overdistention. This phenomenon, associated with elevation of the pulmonary capillary transmural pressure to levels greater than 40 mm Hg, has been called capillary stress failure. The pressure required to produce capillary stress failure is reduced by increased lung volume (49). It may be that lung overdistention results in capillary stress failure and that elevated vascular pressures act in synergy with airway overdistention to produce lung injury. This may be of particular importance in neonatal respiratory failure, where pulmonary hypertension plays such a prominent role and mechanical ventilation may result in alveolar overdistention.
Lung overdistention experiments have been conducted largely on normal animals with initially healthy lungs. The combined effects of lung overdistention and oleic acid injury, inactivation of pulmonary surfactant, or hydrochloric acid injury result in more severe injury than does overdistention or chemical injury alone. This raises the possibility that lung overdistention may be even more harmful to diseased lungs than to healthy lungs.
Work by Gattinoni and associates using computed tomography (CT) in adults with RDS has demonstrated that the lung in RDS has three components (50):
Healthy tissue that has a fairly normal appearance on CT scanning, has essentially normal compliance, and may be overexpanded under the influence of excessive airway pressures and excessive PEEP
Recruitable lung that may be expanded under the influence of variable levels of PEEP
Injured tissue that does not respond to increases in airway pressure
Under conditions of severe lung injury, mechanical ventilation may result in overdistention of the most normal portion of the lung and cause further injury. Recognition of the phenomenon of overdistention lung injury and the possible deleterious effects of mechanical ventilation using high pressures and volumes has resulted in a change in the way that mechanical ventilation is practiced today in comparison with methods used in the recent past. The concept of minimal-excursion ventilation is a direct result of this understanding. Lower tidal volume ventilation and
methods that prevent lung overdistension are now routinely practiced in patients with acute lung injury.
methods that prevent lung overdistension are now routinely practiced in patients with acute lung injury.
Local tissue alkalosis also may play a role in lung injury associated with mechanical ventilation. When a large intrapulmonary shunt is present in respiratory failure, the PCO2 in the ventilated alveoli and their associated capillaries must be far less than 40 mm Hg to achieve a “normal” PaCO2 of 40 mm Hg. The more normal areas of the lung with appropriate matching of ventilation and perfusion must be “hyperventilated” to achieve a normal PaCO2 after mixing with blood of a higher PCO2 that has traversed perfused but unventilated portions of the lung. In theory, the PCO2 in the ventilated areas of the lung could be low enough to result in tissue-damaging alkalosis (51). It is unknown whether this mechanism is clinically important; however, ventilation strategies that do not target normal PaCO2 (permissive hypercapnea) have improved outcomes in acute respiratory failure.
Deleterious Effects of Oxygen
One of the primary functions of the lung is to permit the transfer of molecular oxygen from the respiratory gas mixture to the blood. Supplemental oxygen is added to the respiratory gas mixture to treat respiratory failure when arterial hypoxemia and tissue hypoxia are present. Oxygen is a drug, however, and its pharmacologic properties must be recognized. Oxygen administration may cause direct cellular injury as a result of the increased production of oxygen free radical-containing species that overwhelm the natural protective mechanisms that normally scavenge these highly reactive molecules.
In patients with chronic hypoxia and hypercarbia, ventilatory drive may be unusually dependent on hypoxia. The administration of supplemental oxygen with “normalization” of arterial oxygen levels may depress ventilation and worsen respiratory failure during spontaneous breathing. This phenomenon is seen clinically in older patients with chronic obstructive pulmonary disease, in children with chronic lung disease such as advanced cystic fibrosis, and in infants with bronchopulmonary dysplasia. Oxygen is a pulmonary vasodilator, and is important in the treatment of pulmonary hypertension for this reason. Absorption atelectasis can occur when poorly ventilated areas of the lung contain only oxygen and no inert gases. As oxygen is slowly absorbed, these areas of the lung may become atelectatic. Apart from the lung, chronic oxygen exposure in neonates can result in retinopathy of prematurity. Hyperbaric exposure to oxygen causes CNS injury manifested as seizures and paralysis.
The direct lung injuries associated with oxygen exposure include tracheobronchitis, acute alveolar-capillary injury, and chronic lung injury. A variety of physiologic changes have been described with continuous hyperoxia (52). Early in exposure, mucociliary clearance is decreased. Macrophage phagocytic function is depressed. After only a few hours of breathing 100% oxygen, vital capacity and compliance fall, and adult volunteers uniformly report chest pain. Surfactant production decreases and lung water increases. Vital capacity and diffusing capacity do not return to normal for 2 weeks after 3 days of oxygen exposure. Primates survive for about a week under such exposure. Pathologic changes associated with hyperoxic exposure are seen after a latent period of 24 to 72 hours that varies among species and among individuals. This latent period is followed by a period of acute injury and inflammation, then by a destructive phase. Endothelial cells show injury first. This is followed by type I epithelial cell damage and an increase in the number of type II pneumocytes. Interstitial edema develops, followed by alveolar edema. Neutrophils and monocytes infiltrate the interstitial space coincident with the beginning of cellular injury and, along with resident neutrophils and monocytes, become activated and release cytokines that result in the recruitment of more leukocytes to the lung, and perpetuate the injury.
The highest “safe” concentration of inspired oxygen that can be tolerated indefinitely is unknown. FIO2 concentrations of less than 0.6 result in minimal morphologic changes in rodents. FIO2 of less than 0.5 seem to be tolerated indefinitely by both children and adults. In premature infants, minimal elevations in the FIO2 are associated with retinopathy.
Barotrauma
The subtle morphologic and physiologic changes that can result from overdistention of the lung have been referred to as a form of barotrauma; however, other descriptive names, such as volutrauma, overdistention lung injury, and pressure-volume lung injury, better describe these injuries. The term barotrauma is best used to refer to the appearance of extraalveolar gas as a result of the gross mechanical disruption of the airways. The clinical manifestations of barotrauma include pneumothorax, tension pneumothorax, pneumomediastinum, tension pneumomediastinum, pneumopericardium, pulmonary interstitial emphysema, pneumoperitoneum, pneumoretroperitoneum, subcutaneous emphysema, pneumatocele, bronchopleural fistula, and intravascular air embolism.
When high pressures and corresponding high volumes are created in the lungs, large pressure gradients may be present between the bronchovascular sheaths and adjacent alveoli. Alveoli may rupture into the interstitial tissues of the bronchovascular sheath and allow gas to be introduced. Both high airway pressures and underlying lung disease are usually necessary to cause barotrauma, as evidenced by the fact that adults with normal lungs can sustain seemingly enormous increases in airway pressure without developing extraalveolar air. Coughing, sneezing,
Valsalva maneuver, weight lifting, and playing wind instruments result in airway pressures that may exceed 200 cm H2O. Such normal activities occasionally result in clinically significant barotrauma.
Valsalva maneuver, weight lifting, and playing wind instruments result in airway pressures that may exceed 200 cm H2O. Such normal activities occasionally result in clinically significant barotrauma.
Barotrauma should be regarded as a sign of lung injury associated with the use of mechanical ventilation. Therapy for barotrauma should be directed at the primary disease and consideration given to changing the method of mechanical ventilation. Life-threatening effects of barotrauma may need to be treated immediately. Any pneumothorax that occurs while a patient is on positive-pressure ventilation must be considered a potential tension pneumothorax and should be decompressed by tube thoracostomy. Tube drainage (best performed under ultrasound guidance) of pneumopericardium may be necessary. Ventilator strategies that reduce PEEP, tidal volume, and inspiratory pressure should be used. Permissive hypercapnea may be helpful. Neonates with pulmonary interstitial emphysema may benefit from high-frequency ventilation modes. Pneumoperitoneum always presents a diagnostic dilemma for the surgeon because critically ill patients are at risk for stress ulcers and intestinal ischemia that may result in visceral perforation. Excluding a surgical cause of pneumoperitoneum may be difficult. Contrast studies may be of some benefit, along with serial examinations, in making a decision about the need for laparotomy or laparoscopy.
TREATMENT OF RESPIRATORY INSUFFICIENCY: ASSISTING THE FAILING LUNG
The treatment of respiratory failure must be designed to permit healing of the injured lung, while preserving the remaining uninjured lung. The goals of treatment include improving the inadequate pulmonary gas exchange that results in hypoxemia and acute respiratory acidosis, relieving respiratory distress by decreasing the work of breathing, and altering lung mechanics to improve lung compliance and treat atelectasis (53). The underlying cause of respiratory failure must be treated concurrently.
The deleterious effects of mechanical ventilation, including lung overdistention, oxygen toxicity, and barotrauma are now collectively referred to as ventilator-associated lung injury. Techniques of mechanical ventilation designed to minimize lung injury are referred to as protective ventilation strategies.
General Measures
Position of the Patient
In the mathematic description of the mechanical properties of the lung given earlier, the lung is assumed to be homogeneous. However, the lung is inhomogeneous in both health and disease. Position and gravity play an important role in determining the inflation, distribution of ventilation, and distribution of perfusion of both healthy and sick lungs.
In normal, supine individuals, regional lung inflation decreases exponentially from ventral to dorsal, with the most inflated alveoli near the ventral surface and the most compressed alveoli in the dependent, dorsal regions. In the prone position, regional lung inflation changes; the dorsal portions of the lung are the most inflated and the dependent (ventral) lung is the most compressed. Regional inflation is more homogeneous in the prone than in the supine position. Lung weight and hydrostatic pressure probably play the major role in determining the distribution of lung inflation.
In adults with acute respiratory failure, lung weight may be two or three times that of normal lungs. This lung edema is distributed relatively uniformly throughout the lung tissue and does not accumulate preferentially in dependent regions of the lung (54). Total lung gas volume is markedly decreased, whereas total lung dimensions are unchanged (i.e., interstitial fluid replaces gas volume and FRC is decreased). The rate of decrease in lung inflation along the vertical axis is greater in acute respiratory failure than in normal lungs. In acute respiratory failure, the most dependent regions of the lung are also subject to high hydrostatic pressures from the weight of the homogeneously distributed edema fluid (10 to 15 cm H2O in adults). As in the normal lung, the prone position usually results in greater inflation in the dorsal regions and less inflation in the ventral regions of the lung.
In supine, spontaneously breathing individuals, ventilation is distributed primarily to the dependent portions of the lung. These portions of the lung have a greater capacity to expand because they are relatively more compressed at end expiration. The action of the diaphragm also results in a greater ventilation of the dependent lung. In mechanically ventilated patients, ventilation is preferentially distributed to the nondependent portion of the lung because the dependent regions are more likely to be totally collapsed. In addition, the diaphragm is passive and its upper (ventral) portion faces less intraabdominal pressure.
In the normal lung, blood flow is distributed largely by gravity. In the diseased lung, factors such as vasoconstriction, vessel obliteration, the effect of microemboli, and extrinsic vessel compression play a role in the distribution of blood flow.
Prone positioning can make a dramatic change in the respiratory function of adults with RDS (54,55,56). In supine patients, ventilation goes primarily to the anterior portions of the lung, whereas perfusion goes primarily to the posterior aspects of the lung, creating the ventilation–perfusion mismatch that results in hypoxemia. It seems prudent to use frequent position changes in patients with respiratory
failure. Dependent, compressed portions of the lung may be expanded. Pooling of respiratory secretions may also be prevented. Ventilation–perfusion mismatch may be minimized. Because prone positioning is not practiced routinely in children and adults, and has obvious dangers, side-to-side position changes should be carried out. Diligent nursing is required to ensure that patients are actually turned fully lateral during these position changes. It is difficult to break the habit of routine supine positioning in the ICU. It is noteworthy that nurses who handle newborns with respiratory failure are aware of the position that “the baby likes.” With the instantaneous feedback afforded by continuous pulse oximetry, these fragile infants are positioned empirically in such a way that their oxygenation is optimized. Reports of the benefits of prone positioning in pediatric patients have now appeared (57). A multiinstitutional randomized prospective study is currently underway to assess the effects of prone positioning on pediatric respiratory failure.
failure. Dependent, compressed portions of the lung may be expanded. Pooling of respiratory secretions may also be prevented. Ventilation–perfusion mismatch may be minimized. Because prone positioning is not practiced routinely in children and adults, and has obvious dangers, side-to-side position changes should be carried out. Diligent nursing is required to ensure that patients are actually turned fully lateral during these position changes. It is difficult to break the habit of routine supine positioning in the ICU. It is noteworthy that nurses who handle newborns with respiratory failure are aware of the position that “the baby likes.” With the instantaneous feedback afforded by continuous pulse oximetry, these fragile infants are positioned empirically in such a way that their oxygenation is optimized. Reports of the benefits of prone positioning in pediatric patients have now appeared (57). A multiinstitutional randomized prospective study is currently underway to assess the effects of prone positioning on pediatric respiratory failure.
Heat and Humidification
Any therapeutic gas delivery system should match the normal heat and humidity at the point of entry into the respiratory system. Gas delivered to the nose should have a temperature of 22°C and a relative humidity of 50%, whereas gas delivered to the trachea should be 34°C and have a relative humidity of 100%. During spontaneous respiration, inspired gases are heated and become saturated with water, reaching 100% humidity and 37°C just below the carina. The airway above this point adds heat and humidity to the inspired gas and extracts heat and humidity from the expired gas, acting as a heat and moisture exchanger. When the upper airway is bypassed during endotracheal intubation, excessive heat and moisture are lost if the ventilating gases are not humidified. Heat loss results largely from the heat of vaporization when the inspiratory gases are humidified by the lungs. Air has a relatively low specific heat and the warming of the gases themselves prevents relatively little heat loss. The trachea and upper bronchi are most affected by inadequate humidification of the respiratory gases, with resultant moisture loss. With inadequate humidification, ciliary action is decreased and ultimately lost, mucous glands are damaged, and the mucosa becomes ulcerated. Compliance and surfactant activity decreases, and atelectasis and hypoxia may result. Heating and humidifying the respiratory gases is not an effective method of rewarming a hypothermic patient, but it is important in preventing further heat loss.
Suctioning and Postural Drainage
Gravity can be used to advantage to help clear airway secretions. Specific positions allow dependent drainage of the lobar bronchi. Adjuncts such as vibration and percussion (manual or by mechanical devices) may also help mobilize secretions.
Suctioning is important in the care of intubated patients whose cough mechanisms are disrupted. In nonintubated patients, eliciting a deep cough to clear airway secretions and forcing a vital capacity breath may be facilitated by intratracheal suctioning. Suctioning of the hypopharynx may elicit coughing and clear some secretions. Intratracheal suctioning is accomplished in much the same way as nasotracheal intubation in spontaneously breathing patients. A catheter with a curved tip is passed through the nose and positioned in the hypopharynx. The catheter is manipulated until respirations are audible through it and is then advanced rapidly with inspiration. Proper entry into the trachea is obvious. The use of fiberoptic bronchoscopy allows directed suctioning to be performed, but it is doubtful that it is more helpful than properly performed catheter suctioning in intubated or nonintubated patients.
Prevention of Atelectasis and Incentive Spirometry
Sustained inspiration is the most effective method of preventing and treating atelectasis (58). During normal respiration, deep, sustained breaths are taken in the form of a sigh or yawn. This normal mechanism may not be present in perioperative or critically ill patients and must be encouraged or substituted. Coughing does not promote full lung inflation, whereas a sustained inspiratory effort is effective in fully expanding the lungs. Nonintubated children, able to cooperate, benefit from incentive spirometry. Proper use of an incentive spirometer requires a sustained inspiratory effort. The objective is to create sustained inspiratory effort. Exhalation maneuvers such as blowing bubbles are less effective in lung expansion, but may be performed more easily by younger children. Lung expansion during occasional crying is important for children who are too young to participate in voluntary maneuvers. Pain should be controlled, but oversedation that results in continual somnolence and shallow breathing should be avoided. Intermittent positive-pressure breathing may be beneficial in selected settings.
Intubated patients benefit from intermittent lung expansion to full lung capacity through the use of routine “bagging and suctioning.” Intermittent large tidal volume “sigh” breaths provided by the mechanical ventilator do not really recapitulate the normal mechanisms of full lung inflation and carry a risk of lung injury in diseased lungs. A more physiologic approach to atelectasis treatment and prevention is provided by a sustained breath at 35 to 40 cm H2O that is held for 5 seconds or longer and repeated several times. Vibrating vests and negative pressure assist devices play a role in select cases in treating atelectasis and mobilizing secretions.
Pharmacologic Agents
Antibiotics
When respiratory insufficiency is caused by bacterial infections involving the lung itself (pneumonia), or when bacterial infections involving other organs results in respiratory failure (e.g., necrotizing enterocolitis), antibiotic therapy is used. Appropriate cultures are obtained, including blood, sputum, urine, and cerebrospinal fluid, as indicated by the disease presentation and the age of the child, and broad-spectrum antibiotic coverage is begun. Antibiotic therapy is tailored to specific organisms based on culture results.
Patients who are intubated and receiving mechanical ventilation for any reason are at great risk for acquired bacterial pneumonia; however, prophylactic antibiotics are not indicated when mechanical ventilation is instituted. Gram-negative organisms and Staphylococcus species are the most common infectious agents in intubated patients. The diagnosis of pneumonia and the decision to institute antibiotic therapy in intubated patients can be difficult. Decisions to institute or change antimicrobial therapy should be based on multiple clinical variables, including worsening gas exchange, end-organ signs of sepsis, changes in the chest radiograph, changes in the quantity or quality of respiratory secretions, and the results of respiratory tract cultures. In the face of respiratory deterioration, empiric administration of antibiotics or changing antibiotics may be helpful.
Diuretics and Fluid Management
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


