Renal Physiology and Pathophysiology
John W. Foreman
Delbert R. Wigfall
Department of Pediatrics, Duke University Medical Center, Durham, North Carolina 27710.
Department of Pediatrics, Division of Nephrology, Duke Children’s Hospital and Health Center, Durham, North Carolina 27710.
RENAL PHYSIOLOGY
Renal Blood Flow and Glomerular Filtration
The major role of the kidney is to maintain body water and electrolyte homeostasis. The first step in this process is the production of the glomerular filtrate from the renal plasma. The glomerular filtration rate (GFR) is dependent on renal plasma flow, which in turn is dependent on blood pressure and circulating volume. The kidneys receive about 20% to 30% of the cardiac output, and this is maintained over a wide range of blood pressures through changes in renal vascular resistance. Numerous hormones play a role in this autoregulation, including the vasodilators (prostaglandins E and I2, dopamine, and nitric oxide) and the vasoconstrictors (angiotensin II, thromboxane, adrenergic stimulation, and endothelin). Congestive heart failure and volume contraction severely limit the kidney’s ability to maintain autoregulation in the face of changes in blood pressure.
In neonates, renal blood flow, adjusted for body surface area, doubles during the first 2 weeks of postnatal life and continues to rise until it reaches adult values by the age of 2 years (1,2). This is due to both an increase in cardiac output and a fall in renal vascular resistance. Paralleling these changes in renal blood flow, the GFR when adjusted for body surface area also doubles over the first 2 weeks of postnatal life and continues to rise until it reaches adult values by the age of 1 to 2 years. The initial GFR and the rate of rise correlate directly with gestational age at birth; for example, the GFR of an infant of 28 weeks gestation is one-half that of a full-term infant.
At the level of the glomerular capillary, the GFR is dependent on the balance of the hydrostatic pressure forcing fluid through the capillary wall into Bowman’s space, the oncotic pressure from plasma proteins retarding this flow, the capillary surface area available for filtration, and the permeability of the capillary wall. The net hydrostatic pressure is dependent on the intraglomerular capillary pressure and the pressure in Bowman’s space. Autoregulation maintains intraglomerular capillary pressure over a wide range of systemic blood pressures, although this is blunted by volume contraction. The pressure in Bowman’s space rarely plays a role in determining the GFR, except during some forms of acute renal failure (ARF) with tubular blockage by cellular debris and acute urinary tract obstruction. Changes in plasma oncotic pressure also have little influence on the GFR because these changes are counteracted by changes in the surface area available for filtration.
The whole-kidney GFR is the sum of the GFR of each nephron, of which humans have about 1 million in each kidney. The formation of these nephrons is complete by 36 weeks gestational age. Individual nephrons can alter their GFR in response to several stimuli, including protein loading and nephron loss. In healthy individuals, the GFR can increase by 50% after a large protein meal. Renal donors can increase the GFR in their remaining kidney such that the average donor has a GFR that is 70% of the preoperative value. Children with congenital absence of a kidney have a more marked increase in the size and GFR of the remaining kidney after birth, such that their GFR is comparable to that of their peers with two kidneys. Clinically apparent changes in the GFR are not seen with renal disease until 70% of the nephrons are damaged because the unaffected nephrons increase their individual GFR.
Radionuclides, such as diethylenetriamine pentaacetic acid (DTPA), can be used to measure the GFR accurately and easily. DTPA is freely filtered by the glomerulus and not reabsorbed or secreted by the renal tubule, making it an ideal substance for GFR measurements. One method is to inject a single dose of the radionuclide and plot the fall in radioactivity with timed blood samples. The decline in radioactivity is proportional to the GFR. These GFR
measurements can also be combined with renal imaging studies. The GFR in each kidney can be estimated by determining the radioactivity in each kidney over time.
measurements can also be combined with renal imaging studies. The GFR in each kidney can be estimated by determining the radioactivity in each kidney over time.
Sodium Handling
One of the major roles of the kidney is to regulate total body sodium balance and maintain a normal extracellular and circulating volume (3). The kidneys of a normal adult filter 25,000 mEq of sodium per day, yet excrete less than 1% through extremely efficient reabsorption mechanisms along the nephron. The driving force for most of this sodium reabsorption is the enzyme Na+-K+-adenosine triphosphatase (Na+-K+-ATPase) located on the basolateral membrane. This enzyme exchanges three intracellular sodium ions for two extracellular potassium ions using the energy from the hydrolysis of adenosine triphosphate. This exchange maintains a low (less than 20 mEq per L) intracellular sodium concentration and a negative intracellular-to-extracellular charge. These two forces provide energy to drive sodium ions from the lumen of the tubule into the tubular cell through numerous membrane transporters and channels. These sodium ions are then extruded from the cell, mainly by Na+-K+-ATPase, and move from the renal interstitial space into the peritubular capillary. Movement from the interstitial space into the capillary is dependent on the difference between the tissue and the capillary hydrostatic pressures and the capillary oncotic pressure, especially in the proximal tubule.
The proximal tubule reabsorbs 50% to 70% of the filtered sodium. Sodium enters the proximal tubule cell by the luminal membrane through two general types of transporters. One of these cotransports organic solutes and phosphate, and the other exchanges intracellular hydrogen ion for sodium. Sodium exits the cell mainly through the Na+-K+-ATPase. The descending limb of the loop of Henle does not play a role in sodium balance, whereas the ascending limb, especially the thick portion, reabsorbs about 25% of the filtered sodium. The luminal entry step in this segment is by cotransport with one potassium and two chloride ions (Na+-K+-2Cl– pump). The distal nephron accounts for 10% of overall sodium reabsorption by cotransport with chloride in the distal tubule and through sodium channels in the collecting duct.
The kidney must be able to adapt to marked changes in sodium intake and defend against changes in extracellular volume. Several factors play a role in these adaptations. These changes must be recognized, and it is clear that alterations in both circulating volume and total extracellular volume are “sensed” by the body, although the exact location of all these sensors has not been determined. There is good evidence for sensors in the cardiac atria, carotid sinus, and kidney. There may also be intracranial sensors. Once changes in extracellular volume are recognized, several factors are set in motion to adjust urinary sodium excretion.
One of these factors is a change in the GFR. Increasing dietary sodium intake or intravenous volume expansion increases the GFR, just as volume contraction decreases it. Volume contraction lowers the GFR by increasing renin release, both directly from the kidney and through the renal nerves. This in turn increases the angiotensin II level, which reduces the capillary surface area available for filtration and renal plasma flow. These changes are antagonized by prostaglandins released by the kidney. This reduction in the GFR decreases the amount of sodium filtered. Volume loading has the opposite effect. However, these changes in filtered load are blunted by changes in proximal tubule sodium reabsorption. This has been termed glomerulotubular balance.
Proximal tubule sodium reabsorption is regulated mainly by changes in peritubular capillary hydrostatic and oncotic pressure. Volume depletion leads to an increase in angiotensin II levels that constricts the glomerular arterioles, especially the efferent arteriole. This decreases renal perfusion pressure, renal plasma flow, and the GFR. Renal plasma flow is reduced more than the GFR, increasing the fraction of plasma filtered and the protein concentration of the plasma entering the peritubular capillaries. This increases the capillary oncotic pressure. This increase in capillary oncotic pressure and decrease in capillary hydrostatic pressure promotes the movement of the reabsorbed sodium and water from the interstitial space into the capillary and increases luminal sodium reabsorption. Volume loading has the opposite effect.
In addition to the GFR and physical factors influencing proximal tubule reabsorption, two other factors, the renin–angiotensin–aldosterone system and atrial natriuretic peptide (ANP), play significant roles in regulating sodium excretion. Angiotensin II levels influence the GFR and proximal tubule capillary pressures. Angiotensin II also stimulates the release of aldosterone from the adrenal cortex, and this increases sodium uptake by the cortical collecting duct. ANP is released from the cardiac atria in response to changes in pressure and stretching. ANP increases the GFR by dilating the afferent glomerular arteriole and possibly increasing the capillary surface area available for filtration. ANP also inhibits sodium reabsorption in the inner medullary collecting duct, causing naturesis.
Water Regulation
Serum osmolality is tightly regulated through changes in arginine vasopressin (AVP) release and the appreciation of thirst (4). AVP, also called antidiuretic hormone, is a nonapeptide synthesized in the neurons of the paraventricular and supraoptic regions of the anterior hypothalamus, and transported down their axons into the posterior pituitary, where it is stored and released. Separate cells, osmoreceptors, in the anterior hypothalamus are able to sense changes in plasma osmolality and signal the release of AVP and the sensation of thirst. Plasma osmolality below
280 mOsm per kg (the osmotic threshold) completely or nearly completely inhibits AVP secretion, allowing maximal free water excretion by the kidney. As the plasma osmolality rises above 280 mOsm per kg, AVP secretion rises, leading to an increasingly concentrated urine. As the plasma osmolality reaches 295 mOsm per kg, thirst is sensed. However, these changes in plasma osmolality affect AVP secretion only if they are caused by solutes with limited cellular permeability, such as sodium. Increased plasma osmolality from high levels of urea has little effect on AVP secretion.
280 mOsm per kg (the osmotic threshold) completely or nearly completely inhibits AVP secretion, allowing maximal free water excretion by the kidney. As the plasma osmolality rises above 280 mOsm per kg, AVP secretion rises, leading to an increasingly concentrated urine. As the plasma osmolality reaches 295 mOsm per kg, thirst is sensed. However, these changes in plasma osmolality affect AVP secretion only if they are caused by solutes with limited cellular permeability, such as sodium. Increased plasma osmolality from high levels of urea has little effect on AVP secretion.
AVP is also released in response to a fall in circulating volume or hypotension, although detectable changes in AVP secretion are not observed until the blood volume is reduced by 10% to 15% (5). There is an exponential rise of plasma AVP in response to circulating volume changes, and this rise can markedly exceed the increase in AVP observed as a consequence of osmolality changes. Other stimuli have also been purported to release AVP. Nausea, even in the absence of vomiting, can markedly increase AVP release (6). Less clear are the roles of stress and opiates. Early studies indicated that stress and noxious stimuli could release AVP, but more recent studies do not support this concept. Opiates in subemetic and hypotensive doses actually inhibit AVP release.
AVP decreases free water excretion by the kidney by increasing the water permeability of the collecting duct. Water movement out of the collecting duct is dependent on the establishment of a hypertonic interstitium in the medulla. This occurs through the unique arrangement of water and sodium transport in the loop of Henle. Isosmotic fluid enters the descending limb of the loop. This segment is permeant to water but not sodium, whereas the ascending limb is impermeant to water but actively pumps sodium into the interstitium. The active transport of sodium into the medullary interstitium increases its tonicity and abstracts water from the descending limb, concentrating the luminal sodium in this segment. Sodium then moves passively along its concentration gradient out of the ascending thin limb, adding to the medullary tonicity. This arrangement causes fluid entering the distal tubule to be hypotonic relative to the plasma. This hypotonic fluid then proceeds down the collecting duct, which is impermeant to water in the absence of AVP. When AVP binds to the basolateral membrane of the collecting duct, the cell becomes permeant to water and water moves from the lumen through the cell and into the hypertonic interstitium, where it is removed by the vasa recta. This countercurrent mechanism elaborates both a hypotonic and hypertonic urine. Hypokalemia, hypercalcemia, prostaglandins, lithium, and demeclocycline interfere with urine-concentrating ability.
The ability of neonates to conserve or excrete water is limited compared with older infants (7). Full-term neonates can produce maximally dilute urine with an osmolality of 50 mOsm per kg, whereas premature infants can only dilute urine to an osmolality of 70 mOsm per kg. Despite these relatively low urine osmolalities, the excretion of a water load is prolonged, presumably because of the low GFR. Maximal urine-concentrating ability is also reduced, but reaches adult levels by 4 to 6 months of age. This can be hastened with protein loading, suggesting that some of the decreased concentrating ability is related to the low solute content of infant formula.
Potassium Regulation
In general, the kidney excretes about 10% to 15% of the filtered load of potassium, but under certain conditions, it can excrete more than the filtered load. The proximal tubule reabsorbs about 50% of the filtered potassium load through passive diffusion. Potassium is secreted into the descending limb of the loop of Henle from the medullary interstitium, which has a higher concentration. The ascending limb of the loop of Henle actively reabsorbs potassium from the lumen through the Na+-K+-2Cl– pump. This pump, which can be inhibited by loop diuretics, reduces the potassium content of the tubular fluid entering the distal tubule to 10% of the filtered load. The regulation of potassium excretion occurs in the cortical collecting duct. The principal cell of this nephron segment actively secretes potassium into the lumen, and this secretion is regulated by the intracellular potassium concentration and aldosterone. A potassium-rich diet stimulates aldosterone secretion, which increases potassium excretion. With decreased potassium intake, aldosterone secretion is inhibited and principal cell potassium excretion is stopped; however, another cell type, the intercalated cell, continues to reabsorb potassium, leading to net potassium reabsorption.
Newborns are much less efficient at excreting potassium loads compared with adults (8). The normal range of serum potassium concentrations is higher in newborns. This appears to be related to decreased secretion by the principal cells of the newborn cortical collecting duct. The decreased potassium secretion is not related to decreased aldosterone secretion because these levels are higher in newborns, but rather because the tubular response to aldosterone is blunted.
Renal Acid–Base Regulation
The kidney plays a major role in the day-to-day regulation of acid–base balance and the response to the stress of illness. The kidney must first reclaim 4,000 mEq per day of bicarbonate that is filtered by the glomeruli. Eighty percent to 90% of this occurs in the proximal tubule, with the remaining bicarbonate absorbed in the collecting duct, such that the final urine is virtually free of bicarbonate under usual physiologic circumstances. Bicarbonate ion is reabsorbed through a series of steps involving the secretion of hydrogen ion that combines with filtered HCO3– to form H2CO3. This is rapidly converted into H2O and
CO2 by carbonic anhydrase on the luminal membrane. The CO2 easily traverses the luminal membrane and enters the cell, where it is recombined with H2O to form H2CO3 by intracellular carbonic anhydrase. Carbonic acid rapidly decomposes into H+ and HCO3–. The bicarbonate is exchanged for Cl– across the basolateral membrane. The net effect is the movement of HCO3– from the lumen to the blood. A similar sequence of events occurs in the intercalated cells of the collecting duct, except that they lack luminal membrane carbonic anhydrase. Therefore, the intraluminal breakdown of carbonic acid into CO2 and H2O is much slower and occurs to some extent in the lower urinary tract.
CO2 by carbonic anhydrase on the luminal membrane. The CO2 easily traverses the luminal membrane and enters the cell, where it is recombined with H2O to form H2CO3 by intracellular carbonic anhydrase. Carbonic acid rapidly decomposes into H+ and HCO3–. The bicarbonate is exchanged for Cl– across the basolateral membrane. The net effect is the movement of HCO3– from the lumen to the blood. A similar sequence of events occurs in the intercalated cells of the collecting duct, except that they lack luminal membrane carbonic anhydrase. Therefore, the intraluminal breakdown of carbonic acid into CO2 and H2O is much slower and occurs to some extent in the lower urinary tract.
The next role of the kidney in acid–base balance is the formation of “new” bicarbonate to replace that lost in the neutralization of acid generated by the normal combustion of food, especially protein, and the formation of bone. New bicarbonate is generated within the cells of the distal nephron from the decomposition of H2CO3 formed from H2O and CO2 by carbonic anhydrase, after which it enters the peritubular blood. The H+ generated from the decomposition of this H2CO3 is pumped into the lumen of the collecting duct, where it combines with urinary buffers, chiefly HPO2-4 and NH3, and is excreted. This is called titratable acid. With acidosis, more H+ is pumped, lowering the urine pH to about 4.4 and increasing the amount of acid excreted.
The other means of eliminating H+ is the formation of NH4+. Ammonia is generated in the proximal tubule cell from the catabolism of amino acids, mainly glutamine, and is converted to NH4+. With acidosis, the generation of NH4+ can be increased manyfold and is the principal way of responding to a prolonged acidosis. Hypokalemia also stimulates glutamine catabolism and ammonia generation, whereas hyperkalemia has the opposite effect.
Infants, especially newborns, maintain slightly lower values of pH (7.37) and plasma bicarbonate (22 mEq per L) when compared with older children and adults (pH, 7.39; HCO3–, 24 to 28) (9). They can maintain acid–base homeostasis, but are limited in the ability to respond to an acid load, and this is especially true for prematurely born infants, who in the first 3 weeks of life have only half the ability to excrete an acid load compared with full-term infants (10). The lower plasma HCO3– in infants appears to be the result of a lower plasma threshold concentration of bicarbonate ion at which HCO3– is no longer completely reabsorbed by the kidney. Therefore, infants in the first month of life respond less well to an acid load than do older infants and adults. For example, even high-protein feeding can result in acidosis in the first weeks of life.
Renal Calcium Excretion
About 60% of the plasma calcium is filtered by the glomeruli, of which 98% to 99% is reabsorbed. Sixty percent to 70% of this reabsorption occurs passively in the proximal tubule. Another 20% is reabsorbed in the thick ascending limb of the loop of Henle, which can be inhibited by loop diuretics.
The major site of calcium excretion regulation is the distal tubule, where up to 10% of the filtered load is actively reabsorbed. Parathyroid hormone, vitamin D, thiazide diuretics, and estrogen increase distal calcium reabsorption, whereas acidosis decreases it. Saline loading and glucocorticoids also increase calcium excretion by undefined mechanisms. The cortical collecting duct actively reabsorbs a small fraction of the filtered load, less than 5%. Urinary calcium excretion appears to decrease with age. Normal newborn excrete up to 11 mg per kg per day of calcium (11); whereas older children and adults excrete less than 4 mg per kg per day.
Renal Phosphate Handling
The kidney plays a major role in regulating the plasma concentration of phosphate. Of the filtered phosphate, 99% is reabsorbed by newborns, 90% to 95% by children, and 80% by adults, corresponding to growth rates (12). The renal tubule normally operates at nearly maximal capacity for phosphate reabsorption, especially in infants and children. Increases in dietary phosphate intake lead to increases in phosphate excretion, maintaining plasma levels without necessarily leading to changes in tubular transport. However, reductions in phosphate intake signal the renal tubule to increase tubular transport independently of factors known to influence phosphate transport, such as parathyroid hormone.
Virtually all phosphate reabsorption occurs in the proximal tubule through a sodium cotransport system on the luminal membrane. A small amount is reabsorbed in the more distal nephron. Parathyroid hormone (the most potent factor on phosphate reabsorption), acidosis, and glucagon increase phosphate excretion. Vitamin D, growth hormone, and insulin increase phosphate reabsorption. The higher growth hormone levels found in infants and children may explain their higher phosphate reabsorption rates and, consequently, their higher plasma phosphate levels.
Renal Magnesium Handling
About 97% of the filtered magnesium is reabsorbed by the kidney. About 20% to 30% is reabsorbed in the proximal tubule by passive mechanisms. Another 50% to 60% is reabsorbed in the thick ascending limb of the loop of Henle, and the distal tubule and collecting duct account for another 2% to 5%. No specific hormone has been shown to regulate magnesium excretion, but parathyroid hormone, glucagon, AVP, and calcitonin enhance magnesium reabsorption. Mannitol, furosemide, ethacrynic acid, and, to some extent, thiazides increase magnesium
excretion. Aminoglycosides, cisplatin, and cyclosporine increase magnesium excretion.
excretion. Aminoglycosides, cisplatin, and cyclosporine increase magnesium excretion.
RENAL RESPONSE IN STRESS STATES
To maintain the internal milieu, the kidney must respond to many perturbations to the steady state. These conditions activate renal and extrarenal compensatory mechanisms that attempt to restore homeostasis. In an attempt to maintain the vital body functions, these mechanisms may result in trade-offs. For example, in response to cardiac failure, the kidney retains sodium and water that results in edema.
Hypovolemia
The volume of the extracellular fluid compartment is regulated by control of total body sodium. Changes in the sodium intake result in parallel changes in the plasma volume, which is monitored by sensors of central blood volume and blood pressure. It is the effective blood volume that is detected, and this volume may not always correlate with the actual extracellular fluid volume (Fig. 9-1). This is commonly the case in edema-forming states. In response to hypovolemia, the atrial stretch receptors decrease their rate of firing, which lessens their inhibitory feedback on the cardioinhibitory and vasomotor centers in the brain. As a result, sympathetic nerve activity increases, resulting in vasoconstriction and an elevated heart rate (13). Renal sympathetic nerve activity stimulates renal sodium reabsorption and renin secretion. Intrarenal baroreceptors also stimulate renin secretion. Renin catalyzes angiotensin II formation, which increases renal tubular sodium reabsorption, vasoconstriction of arterioles, and aldosterone secretion from the adrenals. Hypovolemia also increases ACTH (corticotropin) secretion, and ACTH stimulates
aldosterone secretion. Angiotensin II also acts on the central nervous system (CNS) to stimulate AVP and adrenocorticotropic hormone release, thirst, sodium appetite, and sympathetic activity. Hypovolemia stimulates nonosmotic secretion of AVP (5), which increases sodium reabsorption and vasoconstriction. Also in response to hypovolemia, atrial stretch receptors decrease their secretion of ANP. This decreases sodium excretion and increases central venous pressure.
aldosterone secretion. Angiotensin II also acts on the central nervous system (CNS) to stimulate AVP and adrenocorticotropic hormone release, thirst, sodium appetite, and sympathetic activity. Hypovolemia stimulates nonosmotic secretion of AVP (5), which increases sodium reabsorption and vasoconstriction. Also in response to hypovolemia, atrial stretch receptors decrease their secretion of ANP. This decreases sodium excretion and increases central venous pressure.
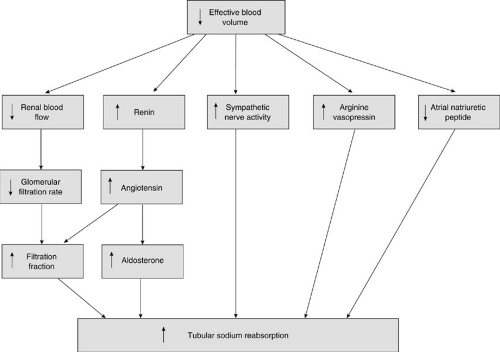 FIGURE 9-1. Activation of renal sodium reabsorption occurs with a decrease in effective blood volume. In response to the activation of a variety of hormonal and nonhormonal factors, the kidney reacts to a decrease in the effective blood volume by increasing renal tubular sodium reabsorption. Vascular responses involve changes in the renal blood flow, glomerular filtration rate, and filtration fraction. Hormonal mediators include arginine vasopressin and the atrial natriuretic peptide. |
These effector mechanisms by which the body protects the integrity of the vascular volume in response to decreased effective plasma volume have potential side effects. The antidiuretic effects of AVP secreted in response to a nonosmotic stimulus contribute to the hyponatremia that is present in many of the conditions associated with decreased effective plasma volume. Potassium wasting and hypokalemia may result from the action of aldosterone on the collecting tubule. Finally, the maintenance of effective blood volume stimulates sodium reabsorption, which may aggravate edema that is already present.
Disturbance of Serum Sodium
Disorders of the serum sodium are not common in children. The symptoms of either hypernatremia or hyponatremia are mainly related to dysfunction of the CNS and include headache, nausea and vomiting, lethargy, disorientation, and even coma. Hypernatremia (serum sodium greater than 145 mEq per L) almost always occurs as a consequence of dehydration and replacement with fluids that contain excess sodium. Rarely, hypernatremia is caused by diabetes insipidus, owing either to an absence of AVP or peripheral unresponsiveness to the hormone. Salt poisoning as a result of child abuse can cause hypernatremia. Correction of the hypernatremia by limiting sodium intake and administering hypotonic fluids should occur over several days to prevent cerebral edema.
Hyponatremia (serum sodium less than 135 mEq per L) can be caused by a number of disorders shown in Table 9-1. Rarely, hyponatremia can be factitious from excess lipids. Hyperglycemia can cause hyponatremia by acting as an osmotic agent attracting water from the ICF and diluting the serum sodium. A 100 mg per dL increase in the serum glucose will decrease the serum sodium by 1.7 mEq per L. The most common cause of hyponatremia in childhood is gastroenteritis-induced volume contraction in conjunction with hypotonic fluid resuscitation. Patients with hyponatremia and normal or mildly expanded total body water often have the inappropriate antidiuretic hormone syndrome (SIADH), usually related to some insult to the CNS or pulmonary disease. These patients have a urine osmolality that is higher than their plasma osmolality and, unlike most patients with hypovolemic hyponatremia, they have a urine sodium concentration that is greater than 20 mEq per L. The third group of hyponatremic patients are those with edema-forming states, such as nephrotic syndrome, congestive heart failure, and cirrhosis. These patients have impaired water excretion from excess antidiuretic hormone. Finally, hyponatremia can also result from excess water intake. In pediatrics, the most common cause of this is the substitution of water for formula because of lack of money to buy formula.
The treatment of hyponatremia depends on the underlying etiology. Hyponatremia associated with volume contraction requires the administration of both sodium and water. The main treatment for SIADH and excess water intake is volume restriction. Volume restriction is also helpful in the edema-forming states along with therapy directed at cause of the edema, such as steroids for nephrotic syndrome. Because of the concern for cerebral edema and neurologic injury, the serum sodium should be corrected slowly in most of these disorders. However, patients with severe symptomatic hyponatremia, such as those with alterations in mental status or seizures, benefit from raising their serum sodium 3 to 7 mEq per L with 3 to 5 mL per kg of 3% hypertonic saline. In general, the rate of serum sodium correction should be limited to ½ mEq per L per hour.
Disturbance of Serum Potassium
Changes in the serum potassium concentration are usually asymptomatic. Both hyperkalemia and hypokalemia can be associated with muscle weakness, electrocardiogram (ECG) changes, and cardiac rhythm disturbances. Factitious hyperkalemia is common in pediatrics and is
usually the consequence of hemolysis during the venipuncture. The average serum potassium decreases with age.
usually the consequence of hemolysis during the venipuncture. The average serum potassium decreases with age.
TABLE 9-1 Causes of Hyponatremia. | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


