Principles of Genetics
Sonja A. Rasmussen
Ada Hamosh
National Center on Birth Defects and Developmental Disabilities, Atlanta, Georgia 30333.
McKusick-Nathans Institute of Genetics Medicine, Johns Hopkins Hospital, Baltimore, Maryland 21287-4922.
Genetic factors contribute significantly to many of the conditions affecting children cared for by pediatric surgeons. Understanding the role of these genetic factors allows for accurate recurrence risk counseling of families, and appreciation of specific genetic disorders can lead the surgeon to identify other manifestations that could affect the patient’s prognosis and surgical management.
Several studies illustrate the contribution of genetic factors to disease in children. For example, about 5% of live-born individuals in a population-based study in British Columbia, Canada, had a condition with an important genetic component that presented before the age of 25 years. The majority of these conditions were classified as multifactorial (46.6/1,000), with fewer classified as single-gene disorders (3.6/1,000) and chromosomal abnormalities (1.8/1,000) (1). A study performed in the early 1970s demonstrated the frequency of genetic conditions among children admitted to a general pediatric hospital; about 25% had a condition with a significant genetic component. Of all admissions, 22.1% had multifactorial/polygenic conditions, 3.9% had single-gene disorders, and 0.6% had chromosome abnormalities (2). Because patients admitted to a pediatric hospital could be viewed as a selected population (possibly biased toward more severely affected patients), a more recent study used population-based data to examine the contribution of birth defects and genetic diseases to all hospitalizations of pediatric patients. In this study, nearly 12% of all pediatric hospitalizations in two states were related to birth defects and genetic diseases (3). These studies emphasize the importance of understanding the principles of genetics and how they relate to conditions treated by pediatric surgeons.
DEOXYRIBONUCLEIC ACID AND GENES
Deoxyribonucleic acid (DNA), the genetic material, is a double helical structure consisting of four bases (adenine, guanine, cytosine, and thymine) on a sugar-phosphate backbone. DNA replication occurs through a semiconservative mechanism; the double helix separates and new strands form using the previous strands as templates. During replication, cytosine will pair only with guanine, and adenine will bind only with thymine. DNA is converted to ribonucleic acid (RNA) by transcription, and RNA is converted to a protein by translation. A set of three bases codes for an amino acid; therefore, the bases make up a code that determines the entire protein’s amino acid sequence. The structure of a gene consists of exons (the portion of the sequence that is translated to protein) and introns, or intervening sequences. After DNA is converted to a primary RNA transcript, the intron sequences, as well as sequences at the beginning and end of the genes, must be removed to form the mature messenger RNA. This message is then transferred from the nucleus to the cytoplasm, where translation occurs.
A recent estimate suggests there are approximately 30,000 to 40,000 genes in the human genome (4). Genes are in pairs, with one member of each pair coming from each parent. The term allele refers to alternative forms of a gene present at a locus, or location on a chromosome. An individual with two of the same alleles at a particular locus is referred to as a homozygote, whereas individuals with two different alleles at a locus are called heterozygotes. When only one allele is present at a locus (such as in loci on the X chromosome in males), the individual is termed hemizygous. The alleles present at a particular locus are referred to as the genotype, and the clinical expression of that genotype in a patient is called the phenotype.
ALTERATIONS IN DEOXYRIBONUCLEIC ACID
Changes in genetic material are called mutations. They can be neutral in effect, disease producing, or rarely protective. Polymorphisms are variations in the DNA sequence that occur at a frequency of more than 1%. These mutations are less frequent in coding regions of the gene and are much more commonly found in introns or between genes. Different types of DNA variations include restriction fragment-length polymorphisms (RFLPs) (5), single nucleotide polymorphisms (SNPs) (6), variable number of tandem repeat polymorphisms (7), and microsatellite markers. SNPs are the most common of these, occurring as often as once in every 100 to 300 base pairs (http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/SNP/). Typically, polymorphisms do not produce disease, although in some cases, a polymorphism can increase a person’s disease susceptibility. Polymorphisms are helpful for following the inheritance of a nearby gene through families, as well as for studying the genetic susceptibility to certain complex diseases (8).
Many different types of disease-producing mutations have been identified (9). A change in a single base pair is referred to as a point mutation. This can be either a missense mutation, in which the base change results in a codon that codes for a different amino acid, or a nonsense mutation, in which the base pair change results in substitution of a stop codon, coding for premature termination of protein translation (but usually causing RNA degradation). Mutations can also alter the initiation codon, and then the initiation of translation does not occur where it should. If a nucleotide changes at or near the intron–exon junction, the splicing mechanism that removes the introns from the coding sequence can be altered. Sometimes, a point mutation occurs such that the resulting codon codes for the same amino acid; this is called a silent mutation. Mutations that change the length of the gene are called insertions and deletions. If the number of nucleotides added or deleted is not a multiple of three, a frameshift mutation occurs. This means that the reading frame for the DNA sequence following the mutation is incorrect, and therefore all the subsequent codons are altered. This generally results in a premature stop codon.
SINGLE-GENE DISORDERS
Single-gene disorders, caused by an error in a single gene, have been catalogued in McKusick’s Mendelian Inheritance in Man since the first edition in 1966. The catalogue is now maintained online, Online Mendelian Inheritance in Man (10), at http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/Omim/. Nearly 1,500 entries were in the first edition, while as of July 18, 2003, the online version had 14,611 entries. The majority of these are autosomal (13,695) with a lesser number being X-linked (813), Y-linked (43), or mitochondrial (60). Online Mendelian Inheritance in Man assigns numbers to single-gene conditions, and these are referred to in this chapter as MIM numbers.
Autosomal Dominant Inheritance
In an autosomal (non–sex-linked) dominant (AD) condition, an abnormality in one member of a gene pair is sufficient to cause the condition. Although the strict definition of a dominant condition specifies that the heterozygous individual is indistinguishable from the homozygous-affected individual, this is often not the case. Instead, frequently the homozygote is more severely affected than the heterozygote.
Most dominant conditions involve the formation of structural proteins. Changes in one gene of a pair can result in a phenotype through loss of function, where the abnormal gene produces an absent gene product. Because only one of the pair of genes is working properly, only half of the normal amount of product is formed. Another way that a dominant gene can cause a genetic condition is through a gain of function, where the new protein takes on a deleterious function (9).
Individuals with a dominant condition have a 50% risk of passing the condition on to their offspring, and males and females are affected with equal frequency. Patients with AD conditions often have other affected family members. Families in which an AD condition is segregating often show a vertical pattern of transmission, with multiple generations affected (Fig. 2-1). Because dominant conditions have a wide variability in expression, a parent might not be identified as having the condition unless he or she is carefully examined for minor manifestations. In some cases, neither parent is affected and the child is a new mutation. The frequency of these new mutations increases with increasing paternal age. Rarely, an individual in an autosomal dominant pedigree who must be a carrier of the gene based on the inheritance pattern shows no manifestations. This is termed incomplete penetrance.
An example of a common AD condition is neurofibromatosis 1 (NF1) (MIM 162200), which occurs in 1 in 3,000 births. This condition is characterized by neurofibromas, benign tumors that arise from the peripheral nervous system. Cutaneous neurofibromas are located just underneath the skin and are primarily of cosmetic significance, whereas plexiform neurofibromas develop from a deeply placed nerve and, because of their size and position, can disrupt growth of other tissues. Other features of NF1, including café-au-lait spots (asymptomatic lightly pigmented macules found on the skin) and Lisch nodules (iris hamartomas that have no effect on vision), are important in establishing the diagnosis. Other features of clinical significance include learning problems or mental retardation, macrocephaly, short stature, scoliosis, hypertension, and increased risk of malignancy (11). The
clinical features in persons with NF1 vary widely; thus, diagnostic criteria have been developed to assist the health care provider in making a diagnosis. NF1 is believed to be fully penetrant, meaning that all persons with an NF1 gene mutation show clinical signs of the condition. Based on clinical data, about one-half of the cases of NF1 are believed to be sporadic mutations, which suggests a high mutation rate for the NF1 gene (12).
clinical features in persons with NF1 vary widely; thus, diagnostic criteria have been developed to assist the health care provider in making a diagnosis. NF1 is believed to be fully penetrant, meaning that all persons with an NF1 gene mutation show clinical signs of the condition. Based on clinical data, about one-half of the cases of NF1 are believed to be sporadic mutations, which suggests a high mutation rate for the NF1 gene (12).
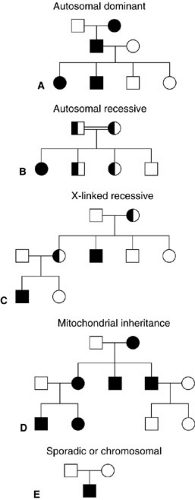 FIGURE 2-1. Various modes of traditional and nontraditional inheritance. Squares denote males; circles are females. Shaded symbols are affected; unshaded are unaffected; half-shaded are carriers. The double line in the autosomal recessive figure (B) signifies consanguinity, which increases the risk of recessive disease. |
This condition is due to mutations in a gene located on chromosome 17 responsible for encoding a protein called neurofibromin. Evidence suggests that one function of the NF1 gene is as a tumor suppressor gene, which if lost or abnormal results in tumor development. This could explain the increased risk of malignancy observed in patients with NF1 (13).
The NF1 gene is very large, spanning approximately 350,000 bases (or 350 kilobases) of DNA, and contains 60 exons (13). The large size of this gene could be responsible for its high mutation rate. Many different types of mutations have been identified, including deletions, insertions, missense mutations, nonsense mutations, and mutations in the introns; only a few mutations have been observed repeatedly in unrelated individuals. Because of this, the specific mutation responsible for the condition has not yet been identified in many patients (14). DNA-based testing in the form of a multistep mutation detection protocol is available (15), but is usually not necessary for diagnosis. In addition, using polymorphisms within the gene, linkage analysis can be used to follow the gene through an affected family (16). More detail about linkage analysis will follow later in this chapter.
Autosomal Recessive Inheritance
In autosomal recessive (AR) conditions, both genes at a locus are abnormal, which results in the absence of normal protein product. Abnormal genes are inherited from both parents each of whom has one normal and one abnormal gene (called carriers), but show no clinical evidence of the condition themselves. Many AR conditions are due to the absence of an enzyme; carriers often have one-half the normal amount of enzyme, but this amount is sufficient so they demonstrate no signs or symptoms of the condition. Two carriers of an AR condition have a 25% risk of having an affected child with each pregnancy (Fig. 2-1). It has been estimated that each of us carries 6 to 10 recessive genes. Therefore, these conditions occur more frequently among inbred groups and consanguineous matings, because related individuals are more likely to have inherited the same abnormal recessive gene.
The finding that some AR conditions are more frequent in particular ethnic groups has led to population screening for carrier status. Some examples of this strategy include carrier screening of the Ashkenazi Jewish population for Tay-Sachs disease and those of Caucasian European or Ashkenazi Jewish descent for cystic fibrosis (CF) (17,18).
An example of a condition inherited in an AR manner is CF (MIM 219700). This condition, occurring in approximately 1 in 3,200 live births in Caucasians, (19) is characterized by chronic pulmonary disease, pancreatic insufficiency, and elevated sweat chloride concentrations. Meconium ileus is seen in 10% to 20% of newborns with CF. The condition is due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on chromosome 7, which codes for a protein that acts as a cell membrane chloride channel. Therefore, patients with this condition have abnormal chloride conduction across the apical membrane of epithelial cells. The most common mutation in patients with CF (accounting for 68% of mutant alleles worldwide, but ranging from 30% to 90%, depending on the ethnic group) (20) results in deletion of a
single amino acid (phenylalanine) from the CFTR protein (termed ΔF508, referring to a deletion of phenylalanine—abbreviated as F—at amino acid no. 508). More than 1,000 other mutations are known, including missense mutations, frameshift mutations (due to addition or deletion of a number of base pairs that is not a multiple of three), nonsense mutations, and mRNA splicing mutations (20).
single amino acid (phenylalanine) from the CFTR protein (termed ΔF508, referring to a deletion of phenylalanine—abbreviated as F—at amino acid no. 508). More than 1,000 other mutations are known, including missense mutations, frameshift mutations (due to addition or deletion of a number of base pairs that is not a multiple of three), nonsense mutations, and mRNA splicing mutations (20).
Evaluations of large numbers of CF patients for the CFTR mutation have allowed study of the correlation between the specific mutation (genotype) and the observed clinical manifestations (phenotype). This correlation has demonstrated that certain CFTR mutations are associated with early onset of pancreatic insufficiency, whereas other mutations have a low frequency of this problem. However, the occurrence of other common complications and the severity and course of pulmonary disease do not appear to be predicted by the CF genotype, but are believed to be due to other genetic and environmental factors (20).
X-linked Recessive Inheritance
In the previously discussed autosomal modes of inheritance, males and females are equally likely to be affected and transmission does not depend on the sex of the parent. This is in contrast to X-linked or sex-linked conditions. Genes for X-linked recessive (XLR) conditions are located on the X chromosome. When a gene is abnormal on one X chromosome in females, the normal gene on the other X chromosome can compensate for the abnormal one. However, a male who inherits an abnormal X is hemizygous, because there is no corresponding gene locus on the Y chromosome, and therefore has the condition. The probability that male offspring of female carriers will be affected is 50%, whereas female offspring have a 50% chance of being a carrier, but are not affected. X-linked inheritance is distinguished from AD inheritance by the absence of male-to-male transmission (Fig. 2-1).
Although most females carrying a mutation for an XLR disorder are asymptomatic, this is not always the case. During embryogenesis in females, one of the X chromosomes undergoes a process known as X-inactivation, or lyonization. This process is random; the maternally and paternally derived X chromosomes have an equal chance of becoming inactivated. However, the same X remains inactivated in daughter cells of the original cell. If by chance, in a large proportion of cells in the involved organ, the normal X chromosome has been turned off, the woman can show symptoms of the XLR disorder.
Hemophilia A (classic hemophilia) (MIM 306700), the most common of the severe congenital coagulation disorders, is inherited in an XLR fashion. Patients with this condition have a reduced coagulant activity of factor VIII in the coagulation cascade, either due to the decreased production of the protein or formation of an abnormal protein that does not work properly. Reduced factor VIII activity leads to recurrent hemarthroses, deep muscle hematomas, intracranial bleeds, and hematuria. Patients are also at an increased risk of bleeding following trauma and surgery. Hemophilias A and B occur in approximately 1 in 5,000 male births, with nearly 80% having hemophilia A (21). In general, the activity of factor VIII in affected males is either absent or severely decreased. Factor VIII activity in female carriers is also reduced, on average, to 50% of normal. However, in about 10% of female carriers, the level of factor VIII activity can be significantly reduced because of inactivation of a high proportion of the normal X chromosomes in the cells of the liver. This can sometimes result in a mild bleeding disorder (22).
Several mutations have been identified in the factor VIII gene. An intron 22 inversion has been observed in 45% of patients with severe hemophilia A (22), whereas an intron 1 inversion accounts for 3% of cases (23). Factor VIII gene deletions or rearrangements, frameshift, splice junction, and nonsense mutations account for about 40% of mutations in severe hemophilia A, with about 10% of mutations being missense. In contrast, the vast majority of mutations in mild to moderately severe hemophilia A (97%) are missense mutations (22). In some cases, the mutation cannot be identified and DNA diagnosis must rely on linkage analysis (discussion follows).
DNA Diagnosis of Single-gene Defects
Advances in the molecular genetics laboratory have led to DNA diagnostic techniques for many single-gene conditions. These methods can be direct or indirect. Direct methods evaluate the DNA for specific mutations. To use a direct method, the gene must have been identified and an efficient technique for identification of the mutation must be available. For example, sickle cell disease is due to a single nucleotide substitution at the sixth codon of the β-globin chain. The mutation changes codon 6 from guanine-adenine-guanine, which codes for glutamic acid, to guanine-thymine-guanine, which codes for valine. Early on, this single nucleotide change was shown to be detectable using a RFLP. The restriction enzyme cuts the normal DNA sequence, but the mutation changes the cut site so it is no longer recognized by the enzyme, allowing diagnosis to be made by examining the size of the resulting DNA fragments (24).
Certain conditions with many different mutations, such as NF1 and hemophilia A (discussed previously), can prove difficult to analyze by direct DNA methods, and thus indirect methods must be employed. Indirect methods are based on the fact that loci located near each other on the same chromosome tend to be inherited together. Markers are chosen that are near to or within the gene of interest. However, these loci can become disassociated by a mechanism called crossing over or recombination, an exchange of genetic material that occurs during meiosis. The
likelihood of the gene and its marker becoming unlinked is directly related to the distance between them. Therefore, polymorphisms are chosen because of their physical proximity to the gene of interest; the closer the gene and polymorphism are, the more likely they will be inherited together (linked). Indirect methods ideally require study of families with multiple affected individuals to allow for determination of phase; that is, which polymorphism is inherited with the normal or abnormal gene. The phase sometimes cannot be determined, and DNA diagnosis in the particular family is then not possible using that polymorphism (the marker is called uninformative). Even when the marker is informative in a family, a cross-over event can unlink the gene and its marker. This means that diagnosis is given as a percentage—if crossing over occurs at a frequency of 5%, then presence of the linked marker accurately detects the disease of concern 95% of the time. However, as more markers are identified, including intragenic markers, the likelihood of an error decreases. Figure 2-2 shows an example of linkage analysis using an intragenic RFLP.
likelihood of the gene and its marker becoming unlinked is directly related to the distance between them. Therefore, polymorphisms are chosen because of their physical proximity to the gene of interest; the closer the gene and polymorphism are, the more likely they will be inherited together (linked). Indirect methods ideally require study of families with multiple affected individuals to allow for determination of phase; that is, which polymorphism is inherited with the normal or abnormal gene. The phase sometimes cannot be determined, and DNA diagnosis in the particular family is then not possible using that polymorphism (the marker is called uninformative). Even when the marker is informative in a family, a cross-over event can unlink the gene and its marker. This means that diagnosis is given as a percentage—if crossing over occurs at a frequency of 5%, then presence of the linked marker accurately detects the disease of concern 95% of the time. However, as more markers are identified, including intragenic markers, the likelihood of an error decreases. Figure 2-2 shows an example of linkage analysis using an intragenic RFLP.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


