Obesity
 |
Because more than one-third of American adults are obese, the unrewarding fight against obesity is all too common, not only with our patients but also with ourselves. An astonishing 64.1% of U.S. women were overweight or obese (28.6% overweight and 35.5% obese) in 2008.1,2 Unfortunately, for over 100 years the incidence of obesity has been increasing in the U.S. and Europe, a reflection of an affluent society with an increasingly sedentary life combined with high caloric foods, but most of the increase has been recent, beginning in 1980.3,4,5 and 6 About 59% of American adults do zero vigorous physical activity in their leisure time.1 This change in lifestyle has produced a high prevalence of obesity with a similar trajectory in adults and children; almost 17% of school-aged children and adolescents in 2008 were obese and 32% overweight.7,8 The only good news is that the prevalence of obesity in the U.S. has not changed since 2003-2004; the upward trend has stopped.2
The lack of success in treating obesity is not due to an unawareness of the implications of obesity; there is a clear-cut, well-recognized relationship between mortality and weight.1 The death rate from diabetes mellitus, for example, is approximately 4 times higher among obese diabetics than among those who control their weight. The death rate from appendicitis is double, presumably from anesthetic and surgical complications. Even the rate of accidents is higher, perhaps because fat people are awkward or because their view of the ground or floor is obstructed. The increase in mortality is not limited to obesity; all overweight individuals have an increase in the risk of death.9 The Nurses’ Health Study estimated that 23% of all deaths in nonsmoking middle-aged women are attributable to being overweight.10
The incidence of hypertension, heart disease, type 2 diabetes mellitus, metabolic syndrome, gout, gallbladder disease, obstructive sleep apnea, osteoarthritis, and all the most
prevalent cancers, including colorectal cancer, endometrial cancer, and postmenopa usal breast cancer, is elevated in overweight people.1,11,12 and 13 Being overweight in adolescenc e is even a more powerful predictor of cardiovascular adverse health effects than being over-weight as an adult.14
prevalent cancers, including colorectal cancer, endometrial cancer, and postmenopa usal breast cancer, is elevated in overweight people.1,11,12 and 13 Being overweight in adolescenc e is even a more powerful predictor of cardiovascular adverse health effects than being over-weight as an adult.14
The increasing prevalence of obesity and its consequences now threaten to replace smoking as the primary cause of preventive mortality. A 40-year-old woman who is a nonsmoker can expect to lose 3.3 years of life if she is overweight (not obese, just overweight)!15 If obese, the loss is 7.1 years, and if smoking is added, the loss is 13.3 years. Unless there is a m ajor change, it is estimated that the rise in U. S. life expectancy experienced over the last several decades will substantially slow down, offsetting the impact of declining smoking.16
When the personal and social problems encountered by obese people are also conside red, it is no wonder that a clinician without a weight problem cannot comprehend why fat i ndividuals remain overweight. The frequency with which a practitioner encounters the obese patient whose weight does not decrease despite a sworn adherence to a limited-cal orie diet makes one question if there is something physiologically different about this pat ient. Is the problem due to lack of discipline and cheating on a diet, or does it also invol ve a pathophysiologic factor? Is the physiology of obese people unusual, or are they simply gluttons? Modern studies of obesity strongly indicate that this is a multifactorial problem, and that lack of willpower and laziness is not the simple answer.
Definition of Obesity
Obesity is an excess storage of triglycerides in adipose cells. There is a difference between obesity and overweight.17 Obesity is an excess of body fat. Overweight is a body weight, including muscle, bone, fat, and body water, in excess of some standard or ideal weight. The ideal weight for any adult is believed to correspond to his or her ideal weight from age 20 to 30. The following formulas give ideal weights in pounds:
Women: 100 + (4 × {height in inches minus 60})
Men: 120 + (4 × {height in inches minus 60})
At a weight close to ideal weight, individuals may be overweight, but not overfat. This is especially true of individuals engaged in regular exercise. An estimate of body fat, therefore, is more meaningful than a measurement of height and weight.
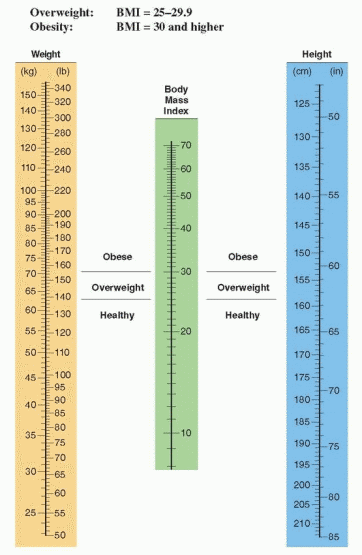
The most accurate method of determining body fat is to determine the density of the body by underwater measurement (hydrodensitometry). It certainly is not practical to measure density by submerging individuals in water in our offices; therefore, skinfold measurements with calipers have become popular as an index of body fat, or expensive imaging techniques can be utilized. These latter methods are not necessary for clinical practice. It is far simpler to utilize the body mass index nomogram, a method that corresponds closely to densitometry measurements.18
The body mass index (the Quetelet index) is the ratio of weight divided by the height squared (in metric units):
To use the nomogram for body mass index (BMI), read the central scale by aligning a straight edge between height and body weight. A body mass index of 25 or more warrants
treatment. Overweight is defined as a BMI of 25 or more (64.1% of American women in 2008). Obesity is defined as a BMI of 30 or more (35.5% of American women in 2008). A “good” BMI for most middle-aged people is in the range of 20 to 24.19 Mortality is lowest in middle-aged women with a body mass index below 19.10,20 A BMI in the overweight or obese range predicts an increased risk for earlier death, no matter the age of the patient.
treatment. Overweight is defined as a BMI of 25 or more (64.1% of American women in 2008). Obesity is defined as a BMI of 30 or more (35.5% of American women in 2008). A “good” BMI for most middle-aged people is in the range of 20 to 24.19 Mortality is lowest in middle-aged women with a body mass index below 19.10,20 A BMI in the overweight or obese range predicts an increased risk for earlier death, no matter the age of the patient.
 |
A person is obese when the amount of adipose tissue is sufficiently high (20% or more over ideal weight) to detrimentally alter biochemical and physiologic functions and to shorten life expectancy. Obesity is associated with four major risk factors for atherosclerosis: hypertension, diabetes, hypercholesterolemia, and hypertriglyceridemia. Overweight individuals have a higher prevalence of hypertension at every age, and the risk of developing hypertension is related to the amount of weight gain after age 25. The two in combination (hypertension and obesity) increase the risk of heart disease, cerebrovascular disease, and death. The Nurses’ Health Study documented a continuing correlation between the body mass index and cardiovascular disease, diabetes, and cancer.10,21,22 In other words, even a modest gain in adult weight, even in a range not considered to be overweight, increases the risk of cardiovascular and metabolic diseases. However, at any given BMI level, the
presence of an increase in abdominal fat, metabolic risk factors, or a strong family history of diabetes, hypertension, and heart disease increases the risk to good health.
presence of an increase in abdominal fat, metabolic risk factors, or a strong family history of diabetes, hypertension, and heart disease increases the risk to good health.
It is well documented that women have a greater prevalence of obesity compared with men. One reason may be the fact that women have a lower metabolic rate than men, even when adjusted for differences in body composition and level of activity.23 Another reason that more women gain weight with age is the postmenopausal loss of the increase in metabolic rate that is associated with the luteal phase of the menstrual cycle. The difference between men and women is even greater in older age.
Unfortunately, the basal metabolic rate decreases with age.24,25 After age 18, the resting metabolic rate declines about 2% per decade. The age-related decline in basal metabolic rate is not observed in women who continue to be involved in a regular endurance exercise program.26 A 30-year-old individual will inevitably gain weight if there is no change in caloric intake or exercise level over the years. The middle-age spread is both a biologic and a psychosociologic phenomenon. It is, therefore, important for both our patients and ourselves to understand adipose tissue and the problem of obesity.
Physiology of Adipose Tissue
Adipose tissue serves three general functions:
Adipose tissue is a storehouse of energy.
Fat serves as a cushion from trauma.
Adipose tissue plays a role in the regulation of body heat.
Each cell of adipose tissue can be regarded as a package of triglyceride, the most concentrated form of stored energy. There are 8 calories/g of triglyceride compared to 1 calorie/g of glycogen. The total store of tissue and fluid carbohydrate in adults (about 300 calories) is inadequate to meet between-meal demands. The storage of energy in fat tissue allows us to do other things besides eating. Our energy balance, therefore, is essentially equivalent to our fat balance. Thus, obesity is a consequence of the fat imbalance inherent in highcalorie diets.
The mechanism for mobilizing energy from fat involves various enzymes and neurohormonal agents. Following ingestion of fat and its breakdown by gastric and pancreatic lipases, absorption of long-chain triglycerides and free fatty acids takes place in the small bowel. Chylomicrons (microscopic particles of fat) transferred through lymph channels into the systemic venous circulation are normally removed by hepatic parenchymal cells where a new lipoprotein is released into the circulation. When this lipoprotein is exposed to adipose tissue, lipolysis takes place through the action of lipoprotein lipase, an enzyme derived from the fat cells themselves. The fatty acids that are released then enter the fat cells where they are reesterified with glycerophosphate into triglycerides. Because alcohol diverts fat from oxidation to storage, body weight is directly correlated with the level of alcohol consumption.27
Glucose serves three important functions:
Glucose supplies carbon atoms in the form of acetyl coenzyme A (acetyl CoA).
Glucose provides hydrogen for reductive steps.
Glucose is the main source of glycerophosphate.
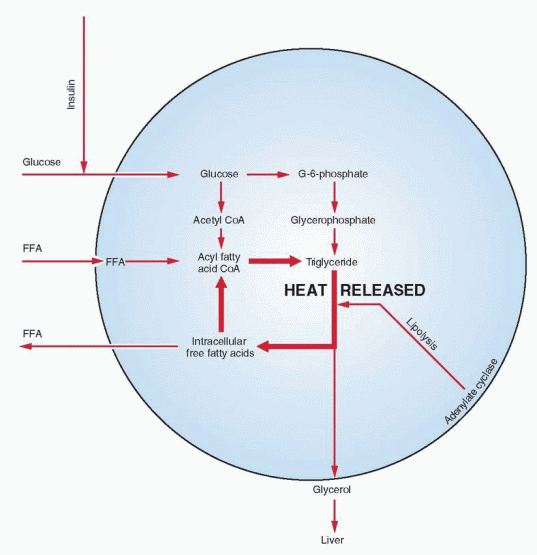 |
The production and availability of glycerophosphate (required for reesterification of fatty acids and their storage as triglycerides) are considered rate limiting in lipogenesis, and this process depends on the presence of glucose.
After esterification, subsequent lipolysis results in the release of fatty acids and glycerol. In the cycle of lipolysis and reesterification, energy is freed as heat. A low variable level of lipolysis takes place continuously; its basic function is to provide body heat.
The chief metabolic products produced from fat are the circulating free fatty acids. Their availability is controlled by adipose tissue cells. When carbohydrate is in short supply, a flood of free fatty acids can be released. The free fatty acids in the peripheral circulation are almost wholly derived from endogenous triglycerides that undergo rapid hydrolysis to yield free fatty acid and glycerol. The glycerol is returned to the liver for resynthesis of glycogen.
Free fatty acid release from adipose tissue is stimulated by physical exercise, fasting, exposure to cold, nervous tension, and anxiety. The release of fatty acids by lipolysis varies from one anatomic site to another. Omental, mesenteric, and subcutaneous fat is more labile and easily mobilized than fat from other sources. Areas from which energy is not easily mobilized are retrobulbar and perirenal fat where the tissue serves a structural function.
Adipose tissue lipase is sensitive to stimulation by both epinephrine and norepinephrine. Other hormones that activate lipase are adrenocorticotropic hormone (ACTH), thyroidstimulating hormone (TSH), growth hormone, thyroxine (T4), 3,5,3′-triiodothyronine (T3), Cortisol, glucagon, as well as vasopressin and human placental lactogen (hPL).
Adipose tissue lipase is sensitive to stimulation by both epinephrine and norepinephrine. Other hormones that activate lipase are adrenocorticotropic hormone (ACTH), thyroidstimulating hormone (TSH), growth hormone, thyroxine (T4), 3,5,3′-triiodothyronine (T3), Cortisol, glucagon, as well as vasopressin and human placental lactogen (hPL).
Lipase enzyme activity is inhibited by insulin, which appears to be alone as the major physiologic antagonist to the array of stimulating agents. When both glucose and insulin are abundant, transport of glucose into fat cells is high, and glycerophosphate production increases to esterify fatty acids.
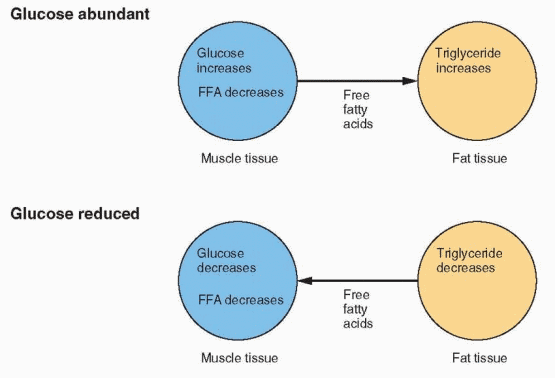 |
The carbohydrate and fat composition of the fuel supply is constantly changing, depending on stresses and demands. Because the central nervous system and some other tissues can utilize only glucose for energy, a homeostatic mechanism for conserving carbohydrate is essential. When glucose is abundant and easily available, it is utilized in adipose tissue for producing glycerophosphate to immobilize fatty acids as triglycerides. The circulating level of free fatty acids in muscle,, therefore, is low, and glucose is used by all of the tissues.
When carbohydrate is scarce, the amount of glucose reaching the fat cells declines, and glycerophosphate production is reduced. The fat cell releases fatty acids, and their circulating levels rise to a point where glycolysis is inhibited. Thus, carbohydrate is spared in those tissues capable of using lipid substrates. If the rise of fatty acids is great enough, the liver is flooded with acetyl CoA. This is converted into ketone bodies, and clinical ketosis results.
In the simplest terms, when a person eats, glucose is available, insulin is secreted, and fat is stored. In starvation, the glucose level falls, insulin secretion decreases, and fat is mobilized.
If only single large meals are consumed, the body learns to convert carbohydrate to fat very quickly. Epidemiologic studies with schoolchildren demonstrated a positive correlation between fewer meals and a greater tendency toward obesity.28 The person who does not eat all day and then stocks up at night is promoting an increase in fat.
Clinical Obesity
Leptin and the Ob Gene (the LEP Gene in Humans)
The hypothalamic location of the appetite center was established in 1940 by the demonstration that bilateral lesions of the ventromedial nucleus produce experimental obesity in rats. Such lesions lead to hyperphagia and decreased physical activity. Interestingly, this pattern is similar to that seen in human beings—the pressure to eat is reinforced by the desire to be physically inactive. The ventromedial nucleus was thought to represent an integrating center for appetite and hunger information. Destruction of the ventromedial nucleus was believed to result in a loss of satiety signals, leading to hyperphagia. Overeating and obesity, however, are not due to ventromedial nucleus damage but rather to destruction of the nearby ventral noradrenergic bundle.29 Hypothalamic noradrenergic terminals are derived from long fibers ascending from hindbrain cell bodies. Lesions of the ventromedial nucleus produced by radiofrequency current fail to cause obesity. These lesions lead to overeating and obesity only when they extend beyond the ventromedial nucleus. Selective destruction of the ventral noradrenergic bundle results in hyperphagia. A sudden onset of hyperphagia can be due to a hypothalamic lesion. Possible causes include tumors, trauma, inflammatory processes, and aneurysms.
Signals arriving at these central nervous system (CNS) centers originate in peripheral tissues. Opiates, substance P, and cholecystokinin play a role in mediating taste, the gatekeeper for feeding, while peptides released from the stomach and intestine act as satiety signals.30 In addition the CNS centers are regulated by locally released neuropeptides. Neuropeptides that inhibit appetite include corticotropin-releasing hormone (CRH), neurotensin, oxytocin, and cyclo(HisPro), a peptide derived by proteolysis of thyrotropin-releasing hormone.31,32 The control of food intake and energy expenditure is very complex, and no agent or system functions in isolation.
The word leptin is derived from the Greek word, “leptos,” which means thin. Leptin is a 167-amino acid peptide secreted in adipose tissue, that circulates in the blood bound to a family of proteins, and acts on the central nervous system neurons that regulate eating behavior and energy balance. Rat studies in the 1950s suggested the existence of a hormone in adipose tissue that regulated body weight through an interaction with the hypothalamus.33,34 But it was not until 1994 that the Ob gene was identified, the gene responsible for obesity in the mouse.35 In the human, this gene is known as the LEP gene.
Genetic Rodent Models of Obesity | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
 |
Ob/ob and db/db mice were described many years ago. The ob/ob mutation arose spontaneously in the Jackson Laboratory mouse colony in 1949. The ob/ob mouse is homozygous for a mutation of the Ob gene on chromosome 6, and the db/db mouse, discovered in 1966, is homozygous for a mutation of the Db gene on chromosome 4.36,37 These mice have been the subject of thousands of publications. The product of the Ob gene is leptin, and in the human, the LEP gene is located on chromosome 7q31.3. Db is the diabetes gene, and this is the locus of the mouse leptin receptor gene. Thus, the ob/ob mouse is obese because it does not produce leptin, and the db mouse is obese because it cannot respond to leptin; its leptin levels are very high (the mutation alters the leptin receptor).
The Leptin Receptor
The leptin receptor belongs to the cytokine receptor family.37 There are multiple isoforms with at least three major forms, a short form and a long form, OB-RS and OB-RL, plus a circulating protein that consists of the extracellular domain. The extracellular domain is very large with 816 amino acids. The intracellular domain of the short form contains 34 amino acids, and in the long form, about 303 amino acids. The short form has many variations, whereas the long form is the common signaling receptor. The only place that the long form is expressed in greater amounts than the short forms is in the hypothalamus, in the arcuate, ventromedial, paraventricular, and dorsomedial nuclei.38,39 High levels of the short-form leptin receptors in the choroid plexus indicate a transport role for the short form from blood into the cerebrospinal fluid to diffuse into the brain.40
The class I cytokine receptor family (to which the long form belongs) acts by proteins that phosphorylate the receptor after binding and STAT proteins that are activated after phosphorylation, and then translocate to the nucleus and stimulate gene transcription. The long-form receptor works through STAT proteins, but gene knockouts specific for STAT proteins are not obese, indicating the presence of other signaling pathways activated by leptin.
The Db gene encodes the leptin receptor. The Db mutation converts the long form to the inactive short form, resulting in leptin resistance and obesity. The db/db mouse has a single G for T nucleotide substitution within the C terminal untranslated end of the short intracellular domain of the ob receptor. This results in a new splice site that creates an abnormal exon inserted into the messenger RNA (mRNA) that encodes the long intracellular domain of the ob receptor. As a result, the long-form mRNA in db/db mice encodes a protein with most of the intracellular domain truncated to be similar to the short form of the ob receptor. The fa/fa mutation is a glutamine for proline substitution in the extracellular domain, and in contrast to the db/db mouse, the fa/fa rat will respond to leptin, but only if it is delivered into the brain.41
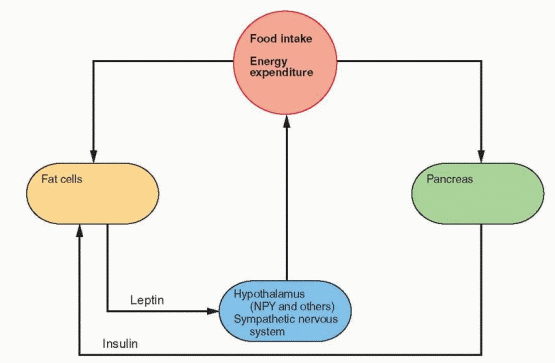
The Physiologic Feedback Loop
Energy expenditure is composed of the basal metabolic rate, diet- and temperature-induced heat production, and the energy necessary for physical activity. Leptin induces weight loss in mice due to decreased appetite and food consumption and an increase in heat production and activity.42 Leptin placed in the lateral ventricle of the rodent brain causes this weight loss, associated with a decrease in the hypothalamic peptide neuropeptide Y (NPY)
expression and secretion. NPY is a 36-amino acid polypeptide that is a potent stimulator of eating when injected directly into the rodent brain.43 NPY has a tyrosine at both ends, hence the use of Y to stand for tyrosine. NPY stimulates food intake, decreases heat production, and increases insulin and Cortisol secretion.
expression and secretion. NPY is a 36-amino acid polypeptide that is a potent stimulator of eating when injected directly into the rodent brain.43 NPY has a tyrosine at both ends, hence the use of Y to stand for tyrosine. NPY stimulates food intake, decreases heat production, and increases insulin and Cortisol secretion.
 |
In rodents, insulin increases the expression of the Ob gene, but there is controversy in humans.42 In humans, there is no surge of leptin after meals, and the acute administration of insulin does not stimulate an increase in leptin levels. However, an increase in LEP gene expression occurs in humans with chronic insulin stimulation, a situation that would be similar to overeating.44 In addition, at least one study has found an acute increase in leptin levels following the induction of hyperinsulinemia (but only in women, not in men).45 It is likely that insulin is at least one regulator of the LEP gene and its secretion of leptin. Thus, this is a physiologic feedback loop to maintain weight and energy.
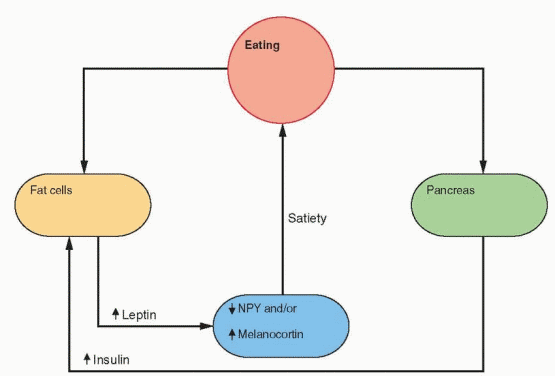
The Ay/Ay rodent becomes fat late in life. The Agouti gene encodes a protein produced by hair follicles. This protein binds to a melanocortin receptor in melanocytes in the skin, thus preventing melanocyte-stimulating hormone action. Agouti rats have high levels of this protein and have yellow instead of black fur. Melanocortins have been demonstrated in the hypothalamus, produced in the arcuate nucleus. Melanocortin binding to its brain receptor influences appetite.46,47 Knockout mice for the melanocortin receptor become obese, and these mice have high NPY expression. Ay/Ay rodents make an excess of Agouti protein, blocking melanocortin action. Thus, too little melanocortin could be another pathway for obesity. The importance of this pathway is evident in the report of mutations within the proopiomelanocortin (POMC) gene characterized by deficiencies in both melanocyte-stimulating hormone (MSH) and ACTH, yielding individuals with obesity, adrenal insufficiency, and red hair pigmentation.48 These findings have culminated in the recognition that mutations in the melanocortin receptor gene account for the most common cause of familial human obesity that can be attributed to a single gene.49
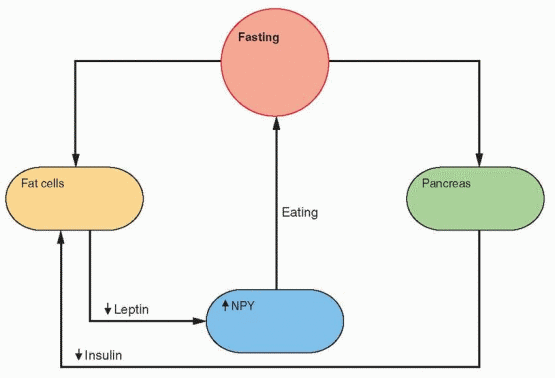
Fasting and exercise decrease leptin secretion and increase NPY gene expression in the arcuate nucleus, followed by release of NPY by neuronal projections into the paraventricular nucleus. The neurons that respond to NPY originate in the arcuate nucleus and project into the paraventricular and dorsomedial nuclei. The arcuate nucleus lies outside the blood-brain barrier (there is no blood-brain barrier in the medial basal hypothalamus) and can be
reached by leptin in the circulation. The NPY neurons stimulate feeding and inhibit heat production by inhibiting sympathetic nervous activity. In ob/ob mice with no leptin, NPY levels are high in the hypothalamus, and leptin treatment lowers NPY and restores everything to normal.38
reached by leptin in the circulation. The NPY neurons stimulate feeding and inhibit heat production by inhibiting sympathetic nervous activity. In ob/ob mice with no leptin, NPY levels are high in the hypothalamus, and leptin treatment lowers NPY and restores everything to normal.38
 |
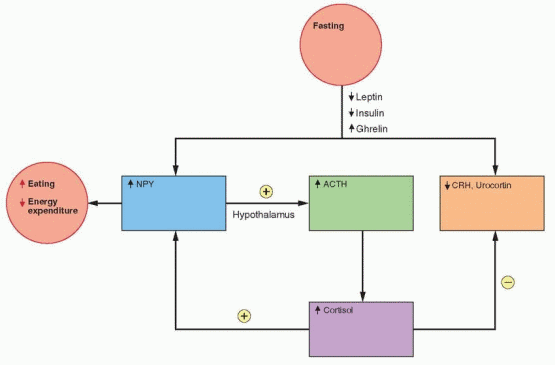
In rodents, caloric restriction increases the expression of NPY in the arcuate nucleus and the release of NPY in the paraventricular nucleus. Leptin and insulin decrease during fasting, allowing an increase in the expression of the NPY gene. Because an increase in metabolism is unwanted during fasting, CRH decreases; however, there is an increase in Cortisol that is a consequence of an NPY-induced hypothalamic signal, not yet identified. Fasting decreases leptin more than expected by the decrease in fat content, indicating the presence of other control mechanisms.50
 |
In normal mice, leptin decreases NPY.51 In most animal models, the NPY content in the hypothalamus is high with obesity, and brain infusions of NPY cause obesity and increased Ob gene expression.52 Knockout mice deficient in NPY maintain normal weight and respond to leptin, indicating that NPY is part of a redundant system, not an absolute requirement.53
 |
In the human, the Ob gene, known as the LEP gene, does not appear to be acutely regulated.54 Feeding increases leptin levels, but slowly, correlating with insulin levels.55,56 Insulin provides negative feedback to the brain in a manner similar to leptin, including an effect on NPY.43 The hyperphagia of diabetes mellitus reflects a deficiency in this insulin action. In fa/fa rats (obese because of a mutation in the leptin receptor), insulin does not affect neuropeptide Y expression, indicating that the inhibition of NPY exerted by insulin is mediated through the leptin-signaling system.57
CRH inhibits food intake and increases energy expenditure; thus, it is expected that weight loss would decrease CRH. In addition, leptin stimulates CRH gene expression (through NPY, POMC, and the melanocortin receptor pathway), and, therefore, lower leptin levels with fasting should lower CRH levels.57 And indeed, CRH secretion is not increased after acute weight loss; however, it is well recognized that Cortisol secretion is increased with stress and exercise. This may be due to a hypothalamic effect of NPY on ACTH secretion by the pituitary.57 The specific food and energy response associated with CRH may also be mediated by urocortin, a CRH-related peptide.58 Urocortin is very potent in reducing food intake, perhaps activating the energy system without activating the overall CRH mechanism.
In summary, circulating levels of leptin provide the hypothalamus with two important pieces of information: how much energy is available, stored in fat, and recognition of acute increases and decreases in energy intake leading to changes in appetite and energy expenditure.
Ghrelin
Ghrelin is a complex hormone, named for its ability to stimulate the release of growth hormone. Ghrelin participates in the regulation of food intake and energy metabolism, but also affects sleep and behavior, inhibits gonadotropin secretion, and is expressed in the ovaries and the placenta.59,60 It influences a wide spectrum of activities, including gastric motility and acid secretion, activity of the pancreas, and the secretion of growth hormone, prolactin, and ACTH.
Ghrelin is a 28-amino acid peptide discovered in 1999 that is secreted mainly in the upper portion of the stomach, but in other tissues as well, including the intestine, pituitary, hypothalamus, kidney, ovary, testis, and placenta.61 The ghrelin circulating in the blood is predominantly from the stomach and intestine, and its target is the energy-regulating centers in the hypothalamus. Given to rodents, ghrelin acutely increases food intake and causes obesity. These actions of ghrelin are independent of its activity in stimulating growth hormone, prolactin, and ACTH secretion, an action that is probably mediated by different ghrelin receptors.62 The targeted neurons are in the same network influenced by leptin (principally the NPY and the endogenous melanocortin receptor antagonist, agouti-related peptide, pathways), with ghrelin and leptin having opposing actions. Another stomach hormone, PYY, acts in a similar fashion as leptin, reducing food intake by modifying the NPY system; this leaves ghrelin as the only hormone known that stimulates food intake.63
The circulating level of ghrelin is lower in obese individuals, reduced with food intake and increased with fasting. Thus ghrelin levels are higher in individuals with anorexia, bulimia, or cachexia; and, in contrast to its appetite-stimulating action in healthy individuals and in patients with cancer cachexia, administration of ghrelin intravenously to young women with anorexia nervosa does not increase appetite.64,65 and 66 These changes are opposite to those of leptin. Ghrelin is a signal to conserve energy by increasing appetite; leptin (and insulin) is a signal to expend energy.64 The key regulator in these responses is the circulating glucose level, but in the case of ghrelin, the system is influenced by meals whereas leptin is modulated by fat mass. The connection between reproduction and the body’s state of energy metabolism is now well established, and the complex leptin-ghrelin system is the means of communication. Specifically, ghrelin exerts a CNS inhibition to prevent reproduction in the presence of an energy deficit, with leptin and ghrelin operating as reciprocal regulators.67
Adiponectin
Adiponectin is another polypeptide secreted by adipose tissue. It influences glucose regulation and fatty acid metabolism; adiponectin circulating levels are relatively high and are inversely correlated with percent body fat.68,69 and 70 Overall adiponectin exerts a weight-losing effect (thus, levels are decreased in obese individuals), complementing the action of leptin in the brain. Adiponectin and its receptors are additional sites where genetic mutations may contribute to obesity. Genetic variants of the adiponectin gene have been described that are associated with the metabolic syndrome and diabetes mellitus.71,72
Leptin in Obese People
Most studies have indicated that nearly all obese individuals have elevated leptin levels, probably due to an increase in LEP gene expression and partly due to greater production because of larger fat cells.73,74 and 75 The greater weight of these individuals represents the leptin level at which stability is achieved, when leptin resistance is overcome. In lean individuals,
leptin levels are similar in men and women, but in women, as weight increases, leptin increases 3 times more rapidly than in men.45 Higher levels in women suggest a greater resistance to leptin, correlating with a greater prevalence of obesity.76
leptin levels are similar in men and women, but in women, as weight increases, leptin increases 3 times more rapidly than in men.45 Higher levels in women suggest a greater resistance to leptin, correlating with a greater prevalence of obesity.76
The incidence of obesity in black and Hispanic women is greater than that of white women.1 Obese postmenopausal black women have 20% lower leptin levels than white women.77 These lower leptin levels correlate with a lower resting metabolic rate in the black women. The lower levels may indicate a greater sensitivity to the leptin mechanism in black women.
In obese individuals, fat cells produce leptin normally and a prevalent genetic failure has not been identified. Thus it is hypothesized that obesity is due to resistance to leptin.78 This may be due to a transport problem of leptin into the brain, as indicated by the finding that leptin level differences between obese and lean individuals are greater in blood than in cerebrospinal fluid.79,80 At least in the mouse, this resistance is present only in the periphery in that leptin administered in the brain still works.81
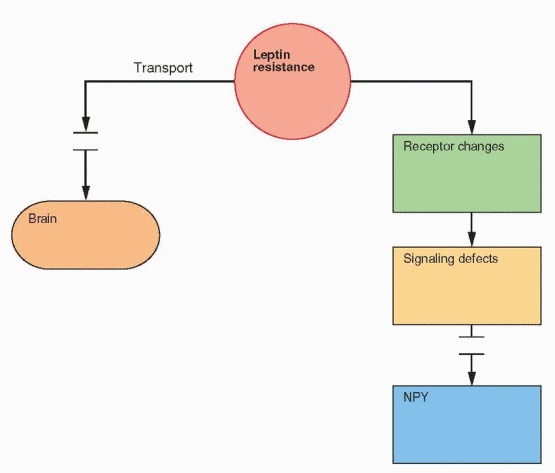 |
In obese individuals, the majority of leptin is unbound and presumably active, consistent with the resistance hypothesis.82 In lean individuals, the majority of circulating leptin is bound. Another important subject for research is the regulation of leptin binding to proteins in the circulation.
Why is Weight Loss so Hard to Maintain?
The average adult eats nearly 1 million calories per year.36 When life was tougher, leptin served the purpose of meeting the threat of starvation. During periods of food availability, individuals with leptin defects could consume large amounts and store excess fat. Today, with no shortage of food, the individuals who survived during food shortages in the past
now become obese, and succumb to the complications of obesity. But the most vexing problem is that 90% to 95% of people who lose weight subsequently regain it.83
now become obese, and succumb to the complications of obesity. But the most vexing problem is that 90% to 95% of people who lose weight subsequently regain it.83
Weight loss in obese and lean people produces the same response, decreased leptin and insulin, and an increase in ghrelin. The obese person tends to regain weight because the lower leptin started from a different set point and is now lower than what the body views as required to be stable in terms of the amount of fat. The ghrelin response also makes it more difficult by stimulating appetite. When energy expenditure, food intake, and body weight are in balance, leptin and ghrelin are at a level consistent with a set point determined by this balance. With weight loss and loss of fat, leptin decreases and ghrelin increases, causing an increase in appetite and a decrease in energy. This is great when you lose weight with an illness, but not good for an overweight person trying to lose weight. Whenever a perturbation occurs, leptin and ghrelin levels change to restore the original status quo, making it difficult for overweight people to maintain weight loss. With both sustained weight loss and persistent weight gain, hormonal mechanisms as well as energy adjustments to maintain lower or higher body weights eventually lead to new set points. However, in the short-term, weight loss can be maintained only by a strict diet or an increase in physical activity to overcome the body’s attempt to restore the original set point.
Circulating levels of leptin are correlated with the percent body fat.73 In other words increased body fat increases the expression of the LEP gene in fat cells. The amount of leptin in the circulation, therefore, is a measure of the amount of adipose tissue in the body. For this reason, neither baseline levels nor initial changes in leptin predict whether weight loss can be maintained.84
A reduction of 10% in body weight is associated with a 53% reduction in serum leptin.73 This would stimulate an effort to regain the weight. The key question is what happens if an individual can successfully maintain the lower weight. In a longitudinal study of obese people, when weight loss was maintained, leptin levels remained low.85
Therefore, whenever a perturbation occurs, leptin and ghrelin levels change to restore the original status quo. Thus, these responses work against attempts to lose weight. A change in energy intake causes a change in leptin levels.57 The body then changes appetite and energy expenditure to conform to the new leptin level. A 10% increase in body weight is associated with a 300% increase in serum leptin.55 The basic purpose is to conserve energy during periods of fasting and to avoid obesity during periods of excess. When caloric intake is reduced, the basal metabolic rate is reduced in a regulatory compensatory adaptation that makes maintenance of weight loss difficult.
Congenital Leptin Deficiency
The LEP gene (Ob gene in mice) has been sequenced from hundreds of obese individuals and mutations are rare.78,86
The first definitive demonstration of a congenital leptin deficiency in humans was reported in two severely obese Pakistani children who were cousins.87 Despite their mass of fat, their serum leptin levels were very low, and the molecular biology study of a fat biopsy revealed a homozygous deletion of a single guanine in the leptin gene. This mutation results in the introduction of 14 aberrant amino acids into the leptin peptide followed by a premature truncation. All four parents were heterozygotes. These children had normal birth weights, but immediately began gaining excessive weight with marked increases in appetites. In contrast to ob/ob mice, they did not have elevated Cortisol levels; however they were hyperinsulinemic. Three obese members of a Turkish family were reported with an amino acid
substitution that impairs intracellular transport of leptin.88 It is noteworthy that these individuals also displayed suppression of gonadal function. Congenital leptin deficiency is now recognized as a rare recessive inherited disorder, the result of mutations at various sites on the LEP gene and associated with hyperphagia and the onset of obesity at an early age.89 Polymorphisms have been described in both the leptin gene and the leptin receptor gene, more prevalent in extremely obese patients, but a relatively low overall prevalence in obese individuals.90
substitution that impairs intracellular transport of leptin.88 It is noteworthy that these individuals also displayed suppression of gonadal function. Congenital leptin deficiency is now recognized as a rare recessive inherited disorder, the result of mutations at various sites on the LEP gene and associated with hyperphagia and the onset of obesity at an early age.89 Polymorphisms have been described in both the leptin gene and the leptin receptor gene, more prevalent in extremely obese patients, but a relatively low overall prevalence in obese individuals.90
With the complexity of the system for energy regulation and the large number of hormones and enzymes involved, we can expect to see multiple mutations identified. An obese individual was reported with a mutation in prohormone convertase (an enzyme that participates in the conversion of prohormones into hormones as does the carboxypeptidase E enzyme deficient in the fat/fat mouse).91 Linkages to obesity have been described to regions near the leptin gene or the leptin receptor gene, perhaps indicating differences in regulatory elements of these genes.92 Remember that melanocortin peptides, along with neuropeptide Y, are part of the leptin signaling pathway in the hypothalamus. Mutations in the melanocortin 4 receptor gene93 are said to be the most frequent monogenetic cause of obesity, yet the 1% or 2% prevalence of this mutation in obese individuals is insufficient to warrant screening.94 It is noteworthy that obese children with melanocortin 4 receptor gene mutations were able to lose weight with lifestyle interventions, but had greater difficulty in maintaining their weight loss.95
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


