Medical and Surgical Complications of Pregnancy
Deborah Krakow
Hematologic Disease
Anemias
Anemia is defined as a hemoglobin (Hb) concentration <12 g/dL in nonpregnant women. Anemia can be either acquired or inherited. During pregnancy, plasma volume expands proportionately more than Hb or red blood cell volume, resulting in Hb dilution, such that anemia is defined as a Hb concentration <10 g/dL. In addition to blood loss, anemia can result from decreased production or increased destruction of red blood cells. The initial workup consists of a history and physical examination as well as an examination of the red blood cell indices and a peripheral smear, with additional tests as indicated (Fig. 17.1).
Iron Deficiency Anemia
Iron deficiency is an acquired anemia and is the most common cause of anemia in gravid women, occurring in 15% to 25% of all pregnancies. Iron deficiency is suspected when the mean corpuscular volume (MCV) is <ã80/mm3 and is confirmed by demonstrating an elevated total iron-binding capacity (TIBC), a low serum iron level, a serum iron-to-TIBC ratio <20%, or a low ferritin level. Effects of iron deficiency on the fetus are usually minimal, although the incidence of neonatal anemia is increased. Iron is transported actively across the placenta, and fetal iron and ferritin levels are three times higher than maternal levels. While mild anemia is not a significant risk factor, severe anemia (Hb <8 g/dL) is associated with intrauterine growth restriction (IUGR). In pregnant women, iron deficiency can cause symptoms including fatigue, headache, lightheadedness, and reduced exercise tolerance. Blood loss at delivery may be tolerated poorly in anemic patients, and postpartum tissue healing may be compromised. For these reasons, treatment during pregnancy is recommended.
The total iron requirement of pregnancy is 1,000 mg: 500 mg increases the maternal red blood cell mass, 300 mg is transported to the fetus and placenta, and 200 mg compensates for blood loss at delivery. The iron requirements of pregnancy increase steadily toward term but average 3.5 mg per day. Even though iron absorption efficiency increases during pregnancy, excess iron must be ingested to ensure sufficient dosage. Recommended supplementation for nonanemic gravidas is 300 mg of ferrous sulfate per day, which contains 60 mg of elemental iron. Anemic gravidas (Hb of 8 or 9 g/dL) should take 300 mg ferrous sulfate two or three times per day. Patients who cannot tolerate iron tablets may take an enteric-coated tablet or a liquid suspension (Table 17.1). Vitamin C facilitates iron absorption. Therapeutic results can be expected after 3 weeks of therapy.
The severely anemic patient (Hb <8 g/dL) may require parenteral therapy in the form of intramuscular or intravenous iron dextran. Because 0.2% to 0.3% of patients have an anaphylactic response to iron dextran, all patients should receive a small test dose 1 hour before the initiation of treatment, and therapy should be provided in an area with ready access to resuscitative medication and equipment. Adequate parenteral therapy should result in
a marked increase in the reticulocyte count within 7 to 14 days.
a marked increase in the reticulocyte count within 7 to 14 days.
 Figure 17.1 Workup of anemia in pregnancy. (MCV, mean corpuscular volume; Hct, hematocrit; Hg, hemoglobin; TIBC, total iron-binding capacity; Fe, iron; HgA2, hemoglobin A2; RBC, red blood cell; G6PD, glucose-6-phosphate dehydrogenase.) |
Megaloblastic Anemia
Megaloblastic anemia is characterized by red blood cells with increased MCV and white blood cells with altered morphology (hypersegmented neutrophils, anisocytosis, and poikilocytosis). It complicates up to 1% of pregnancies and usually is caused by folate deficiency, although it can occur after exposure to sulfa drugs or hydroxyurea or, rarely, because of vitamin B12 deficiency.
Folate deficiency can develop over a relatively short time, as liver stores of folate are sufficient to meet the body’s needs for only 1 to 2 months. Malnutrition (e.g., alcoholism), malabsorption, anticonvulsant therapy, oral contraceptive use, or pregnancy can rapidly deplete the body’s folate stores. Hypersegmented neutrophils (more than 5% of neutrophils having five or more lobes) appear after 7 weeks of deficiency, red blood cell folate is reduced after 18 weeks, and anemia occurs after 20 weeks. The daily folate requirement for a nonpregnant individual is 50 to 100 mg; a pregnant woman needs 300 to 400 mg. This dosage may be difficult to achieve through dietary manipulation because folate is found primarily in fresh fruits and vegetables and is destroyed by cooking. In addition, some individuals need excess folate to prevent neural tube defects. For these reasons, women who are contemplating pregnancy should be advised to ingest a daily folic acid supplement (0.4 mg per day if there is no family history of neural tube defects; 4.0 mg per day if there is a family history) beginning before conception and continuing throughout the first trimester of pregnancy.
TABLE 17.1 Iron Preparations and Dosages | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In contrast, vitamin B12 deficiency is rare because very little of the body’s stores are used each day. Ingested vitamin B12 is bound to an intrinsic factor produced by the parietal cells of the stomach and then absorbed through the mucosa of the distal ileum. Patients who have had a gastrectomy, ileitis, or ileal resection or who have pernicious anemia, pancreatic insufficiency, or intestinal parasites eventually may develop vitamin B12 deficiency.
When megaloblastic anemia is suspected, the history should be reviewed for predisposing factors. The peripheral smear should be examined both to confirm altered cell
morphology and to rule out a mixed (i.e., folate and iron) deficiency. Serum folate and vitamin B12 levels should be measured. A fasting folate level <3 ng/mL or a vitamin B12 level <80 pg/mL indicates deficiency. Folate deficiency responds to 0.5 to 1.0 mg folate orally per day, while a B12 deficiency requires vitamin B12, 1 mg intramuscularly, weekly for 6 weeks.
morphology and to rule out a mixed (i.e., folate and iron) deficiency. Serum folate and vitamin B12 levels should be measured. A fasting folate level <3 ng/mL or a vitamin B12 level <80 pg/mL indicates deficiency. Folate deficiency responds to 0.5 to 1.0 mg folate orally per day, while a B12 deficiency requires vitamin B12, 1 mg intramuscularly, weekly for 6 weeks.
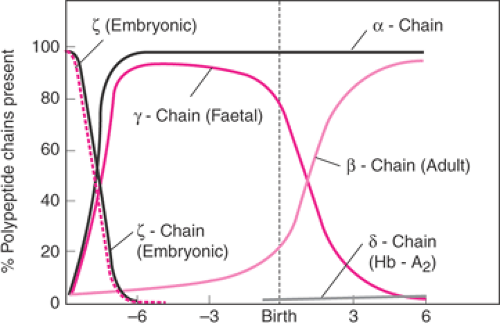 Figure 17.2 roduction of Hb polypeptide chains in relationship to gestational age. (From Rucknagel DL, Laros RK. Hemoglobinopathies: genetics and implications for studies of human reproduction. Clin Obstet Gynecol 1969;12:4 , with permission.) |
Hereditary Anemias
The most commonly encountered hereditary anemias in pregnancy are the thalassemias and sickle cell variants. Hb is a tetramer composed of two copies each of two different polypeptide chains; the identity of the chains determines the type of Hb produced. During embryonic and fetal life, genes directing production of different types of polypeptide chains, and thus different types of Hb, are switched on and then off sequentially. At birth, a normal individual produces α and β chains, along with very small quantities of other Hbs (Fig. 17.2). Normal adults primarily produce hemoglobin A (HbA), composed of two α and two β polypeptide chains.
TABLE 17.2 Hemoglobin Electrophoresis Findings in Various Hemoglobinopathies | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Thalassemias
Thalassemias are characterized by impaired production of one or more of the peptide chains. Thalassemia has a high incidence in certain ethnic groups, especially those originating in the Mediterranean basin, the Middle East, Africa, Asia, and India. Four clinical syndromes are associated with α-thalassemia, and two syndromes are associated with β-thalassemia.
Two genes direct β-chain production, both on chromosome 11. Over 100 different gene mutations have been identified that either prevent or diminish β-chain transcription; if an individual carries an abnormal allele, then β-chain production will be reduced by one half, and abnormally low quantities of Hb will be produced. This results in β-thalassemia minor. The excess α chains combine, instead, with δ chains, producing a molecule called hemoglobin A (HbA2), or with ν chains, producing fetal hemoglobin (HbF). If β-thalassemia minor is suspected because the patient has microcytic anemia without iron deficiency, Hb electrophoresis should be performed. Levels of HbA2 >3.5% and HbF ≥2% confirm the diagnosis (Table 17.2). The gravid patient with β-thalassemia minor generally tolerates pregnancy well. She should receive folic acid supplementation but not iron supplementation unless iron deficiency also is diagnosed. Patients with mutations preventing transcription of both β-chain genes have β-thalassemia major (β-thalassemia), or Cooley anemia. Erythropoiesis is ineffective because there is no β-chain production, and resultant α chains precipitate, causing red blood cell destruction. Occasionally, the mutations allow some β-chain production, resulting in a less severe reduction of Hb synthesis. Aggressive intervention in infancy by using transfusion therapy ultimately leads to iron overload and hemosiderosis, with multiple organ system dysfunction and infertility. Increasing numbers of pregnancies in this population are being reported by virtue of aggressive transfusion and iron chelation therapy. Such pregnancies can be complicated by an increased risk of cardiac arrhythmias and congestive heart failure secondary to severe anemia, chronic hypoxemia, and myocardial hemosiderosis. The safety of iron-chelating agents such as deferoxamine
has not been established in pregnancy and in theory could have an effect on fetal bone development. Prenatal diagnosis is available, and such pregnancies show improved outcome with stable maternal disease.
has not been established in pregnancy and in theory could have an effect on fetal bone development. Prenatal diagnosis is available, and such pregnancies show improved outcome with stable maternal disease.
Before assessing whether a fetus has thalassemia major when the mother is a carrier of thalassemia minor, the father of the fetus should be offered testing. If the father has a normal Hb electrophoresis, the fetus has a 50% chance of having β-thalassemia minor and a 50% chance of being unaffected. If the father has β-thalassemia minor, the fetus has a 25% chance of having β-thalassemia major, a 50% chance of thalassemia minor, and a 25% chance of having normal Hb. Prenatal testing of the fetus should be offered to high-risk couples. Alpha-chain production is directed by four genes, each residing as pairs on chromosome 16. Mutation in only one of the genes results in no clinical or laboratory abnormalities and is thus referred to as the silent carrier state. Mutations in two of the four genes results in β-thalassemia minor, a condition characterized by mild microcytic hypochromic anemia. These patients have a low MCV but normal levels of HbA2. The patient with these laboratory results should be referred for genetic evaluation and family studies to confirm the diagnosis, but typically they tolerate pregnancy fairly well.
Mutation of three of the four α genes results in hemoglobin H (HbH) disease. Affected patients have some HbA and a large percentage of HbH (four β chains). The clinical course is characterized by chronic hemolytic anemia that may worsen during pregnancy. Loss of all four β-chain genes causes β-thalassemia major, resulting in fetal hydrops and perinatal death. As with β-thalassemia, testing of the father is crucial for accurate genetic counseling. Consideration of the patient’s ethnic background also is important. Asians with β-thalassemia minor usually have the two mutant genes on the same chromosome (cis position) and thus have a 50% risk of passing on both affected genes with each conception. In contrast, patients of other ethnic origins usually carry the mutant genes on opposite chromosomes (trans position), so only one affected gene can be transmitted with each conception. All forms of β-thalassemia can be diagnosed by prenatal invasive methods and should be offered to at-risk couples.
Hemoglobinopathies
Hemoglobinopathies involve mutations in the genes encoding Hb. These Hb variants generally have either reduced oxygen transport capabilities or produce hemolytic anemia. Hemoglobin S (HbS) and hemoglobin C (HbC) are the most frequent variants, and they can occur in association with thalassemia as well (Table 17.3).
Sickle Cell Disease
A mutation leading to a single amino acid substitution of valine for glutamic acid at the sixth amino acid residue on the β chain protein changes normal Hb to sickle Hb. An individual who is homozygous for this mutation has sickle cell anemia, or sickle cell disease, producing only HbS and a small quantity of HbF but no HbA. Sickle Hb functions well in the oxygenated state but aggregates, forming rod-shaped polymers, in the deoxygenated state. Polymerized Hb precipitates in the red blood cell, changing the cell from a biconcave disc to an elongated crescent or sickle shape. Sickled red blood cells are not deformable and cannot squeeze through the microcirculation. Microvascular obstruction results in local hypoxia that leads to a vicious cycle of further sickling and obstruction. Localized ischemia and infarction cause tissue damage.
TABLE 17.3 Frequency of Sickle Hemoglobinopathies in Blacks | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Patients with sickle cell anemia usually produce increased quantities of HbF. HbF is not distributed uniformly among all red blood cells but is present at levels of 0% to 20% per cell. In cells containing HbF, restoration of normal oxygen tension may reverse the sickling and halt the destructive process. Cells containing little or no HbF become irreversibly sickled and are rapidly cleared from the system in a process leading to hemolytic anemia. Patients with predominant HbS typically have hematocrits of 20% to 30% and reticulocyte counts of 10% to 25%. Hydroxyurea therapy has been shown to increase both the number of red blood cells containing HbF and the quantity of HbF per cell. However, the issues surrounding the use of hydroxyurea are controversial; not all patients respond to treatment, there are associated complications, and its safety in pregnancy has not been established. It is a known teratogen, although isolated case reports on exposure in humans have been associated with unaffected fetuses. There has been increasing interest in the use of bone marrow/stem cell treatment for this disease. Any pathologic state causing acidosis, dehydration, or hypoxemia can precipitate sickling, hemolysis, vasoocclusion, and infarction. Pregnancy often is characterized by an increase in sickle crises and associated complications (e.g., pneumonia, pyelonephritis, pulmonary emboli, congestive heart failure) and by pregnancy complications (e.g., IUGR, preterm birth, preeclampsia). The goal of pregnancy management should be to maintain adequate hydration and oxygen delivery to the tissues and to avoid or rapidly control infections or other stressors that could precipitate a crisis.
Patients with sickle cell anemia should ingest 1 mg of folate per day to support increased erythropoiesis in the
face of chronic hemolysis and should receive the polyvalent pneumococcal vaccine because chronic splenic infarction leads to functional asplenia by adulthood. Iron supplementation should not be given prophylactically but should be prescribed if there is laboratory evidence of iron-deficiency anemia. All sickle cell patients should undergo a funduscopic examination, with laser therapy as needed, because they are at increased risk for proliferative retinopathy. Asymptomatic bacteriuria (ASB) and other infections should be treated aggressively.
face of chronic hemolysis and should receive the polyvalent pneumococcal vaccine because chronic splenic infarction leads to functional asplenia by adulthood. Iron supplementation should not be given prophylactically but should be prescribed if there is laboratory evidence of iron-deficiency anemia. All sickle cell patients should undergo a funduscopic examination, with laser therapy as needed, because they are at increased risk for proliferative retinopathy. Asymptomatic bacteriuria (ASB) and other infections should be treated aggressively.
One controversy concerns the possible benefit of antepartum prophylactic exchange transfusions. Available data indicate that although some women with sickle cell anemia may escape prophylactic transfusion because they have no associated organ damage, very few crises, and a high percentage of HbF, many will require antepartum transfusion. Even those patients who avoid transfusion during pregnancy should be transfused prior to delivery, because the stresses of labor, anesthesia, operative delivery, and any associated complications (e.g., preeclampsia, chorioamnionitis) can precipitate a serious crisis. Transfusions should be planned to achieve a hematocrit above 30% and an HbA above 50%, although lower values are advocated by some authors. Unless complications dictate otherwise, delivery can be at term, with cesarean section for obstetric indications only.
Substitution of lysine for glutamic acid at the sixth amino acid position of the Hb chain results in the production of HbC. HbC is less soluble than HbA and can cause a mild hemolytic anemia, but it is more stable than HbS under hypoxic conditions. Nonpregnant women who are compound homozygotes for HbS and HbC generally have less severe anemia and fewer pain crises than women with sickle cell disease (HbSS), but under the stress of pregnancy, they experience similar maternal morbidity and pregnancy complications. Additionally, severe bone pain frequently occurs in individuals with hemoglobin SC disease (HbSC), and acute respiratory compromise as the result of embolization of necrotic bone marrow has been reported. The antenatal management of women with HbSC should be comparable to that of women with HbSS.
If one β-chain gene carries the sickle cell mutation and the other gene is functionally deleted, the patient has sickle cell β-thalassemia. Pregnancy-related morbidity in these patients is the same as for sickle cell anemia, and they should be managed similarly. Patients with hemoglobin C disease (HbCC) or C-β-thalassemia have a very mild anemia and usually do not experience hemoglobinopathy-related pregnancy complications.
Heterozygotes for the sickle Hb mutation are referred to as having sickle cell trait (HbSA). Individuals with HbSA have red blood cells that sickle under conditions of markedly reduced oxygen tension. Hb electrophoresis confirms the presence of 55% to 60% HbA in addition to 35% to 40% HbS. Sickling does not occur in vivo, except under conditions of severe stress and hypoxia. Because the renal medulla is especially sensitive to reduced oxygen tension, patients with HbSA may have episodes of painless, self-limited hematuria. During pregnancy, HbSA individuals exhibit an increased susceptibility to urinary tract infections. Patients with HbSA should be offered genetic counseling, and the father of the fetus should be tested so that the precise risk to the fetus can be provided. Prenatal diagnosis is possible by direct DNA analysis by chorionic villus sampling (CVS) or amniocentesis. Generally, no special therapy is required during labor and delivery for these patients.
Congenital Hemolytic Anemias
Hereditary Spherocytosis, Elliptocytosis, and Pyropoikilocytosis
Hemolytic anemia can occur for a variety of reasons. It may result from a hemoglobinopathy; may be autoimmune, drug induced, or pregnancy induced (very rarely); or may occur as the result of inherited red blood cell membrane abnormalities. Hereditary spherocytosis, elliptocytosis, and pyropoikilocytosis result from congenital defects of different red blood cell membrane proteins. All are autosomal dominant disorders occurring at an incidence of 1 in 4,000 to 5,000 in the general population. All result in variant red blood cell shapes, such that affected red blood cells cannot pass readily through the spleen. While trapped in the spleen, the cell membranes are damaged, leading to red blood cell lysis, hemolytic anemia, jaundice, and splenomegaly. Splenectomy is the treatment of choice and effectively eliminates the anemia. Most women of reproductive age with these disorders will already have undergone splenectomy. Although the abnormality of red blood cell shape persists, affected women tolerate pregnancy, labor, and delivery well, with few associated problems. The rare patient who has not undergone splenectomy may experience hemolytic anemia sufficient to require red blood cell transfusions. All patients should receive the polyvalent pneumococcal vaccine and should ingest a folic acid supplement throughout pregnancy. Infection should be treated aggressively, as it may cause hemolysis. The offspring of affected individuals have a 50% chance of inheriting the condition. Affected neonates may experience severe neonatal jaundice requiring exchange transfusion or splenectomy.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited disorder caused by a defect of an enzyme essential to the hexose monophosphate shunt. Because of this defect, when under oxidant stress, Hb sulfhydryl groups become oxidized and Hb precipitates in the red blood cell, leading to hemolytic anemia. The gene is most prevalent among individuals of African, Asian, Mediterranean, or Middle Eastern origin. Known stressors include viruses; bacteria; toxins; fava beans; and certain drugs such as antimalarial agents, sulfa drugs, and nitrofurantoin. Over 400 different gene mutations leading to G6PD deficiency have
been described; the A variant is most common and is present in 1 in 20 black men and 1 in 10 black women in the United States. Although G6PD deficiency is X-linked and males are affected preferentially, carrier females with this gene defect can be symptomatic. Some females have markedly reduced G6PD levels because of unfavorable lyonization, and homozygosity for G6PD deficiency in females can occur (in at least 1 in 400 black women). Precipitating drugs should be avoided in known carriers.
been described; the A variant is most common and is present in 1 in 20 black men and 1 in 10 black women in the United States. Although G6PD deficiency is X-linked and males are affected preferentially, carrier females with this gene defect can be symptomatic. Some females have markedly reduced G6PD levels because of unfavorable lyonization, and homozygosity for G6PD deficiency in females can occur (in at least 1 in 400 black women). Precipitating drugs should be avoided in known carriers.
Platelet Disorders
Thrombocytopenia, defined as a platelet count <150,000 platelets/mm3, occurs relatively frequently in pregnancy, complicating 7% to 8% of all pregnancies. The diagnosis of benign or essential gestational thrombocytopenia is one of exclusion, however, requiring that other pathologic forms of thrombocytopenia be ruled out. Thrombocytopenia in pregnancy can be caused by defective platelet production (bone marrow pathology such as leukemia, lymphoma, metastatic disease), sequestration (splenomegaly), or accelerated platelet destruction. Accelerated destruction is the most common mechanism of disease. Destructive processes unique to pregnancy (e.g., preeclampsia, placental abruption) may occur as part of sepsis or disseminated intravascular coagulation or may result from immunologic dysfunction (e.g., systemic lupus erythematosus, immune thrombocytopenic purpura). These causes of thrombocytopenia are discussed in other areas of the text.
Thrombotic Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura (TTP) is a disorder characterized by the pentad of thrombocytopenia, hemolytic anemia, fever, neurologic abnormalities, and renal failure. It is rare and usually of unknown etiology; however, a rare congenital form caused by mutations in the ADAMTS13 gene that encodes the von Willebrand factor (vWF) cleaving protease have been reported. TTP affects individuals of all ages, although most commonly young women. The untreated mortality rate exceeds 90%. Patients typically experience bleeding (uterine, gastrointestinal, or other) along with a mild Coombs-negative hemolytic anemia, thrombocytopenia, and mild jaundice. Hypertension and renal failure occur later in the course of the disease. All disease signs and symptoms result from microvascular damage caused by platelet thrombi, fibrin deposition, and microaneurysms in arterioles. Endothelial cell function, including prostaglandin production, is abnormal, although it is not known whether this causes TTP or results from it. Immune dysfunction may play a role.
When TTP manifests in the third trimester, it may be difficult to distinguish from preeclampsia or the syndrome of hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). One distinguishing feature is that tests of coagulation (prothrombin time, partial thromboplastin time, fibrinogen, fibrin dimers) usually have normal results in TTP. The advent of a fever of unknown origin or transient neurologic symptoms, as well as nonspecific complaints of arthralgias, nausea, or abdominal pain, may aid in the diagnosis of TTP. End-organ damage worsens as the disease persists. Delirium, seizures, hemiparesis, visual field defects, and coma indicate a very poor prognosis and an increased risk of mortality.
Distinguishing TTP from preeclampsia in its various forms is vital, because management is dramatically different. TTP responds only to plasmapheresis or exchange transfusion, although delivery is eventually curative for preeclampsia. Steroids, heparin, splenectomy, and antiplatelet drugs have had only variable success in the management of TTP. Plasmapheresis should be initiated as soon as the diagnosis is made, regardless of the clinical severity. If the patient is at or near term, magnesium sulfate therapy and delivery also should be initiated because of the possibility that the actual diagnosis is preeclampsia. Cesarean delivery should be for obstetric indications only.
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) is similar to TTP, with similar microangiopathy, except that the kidneys are primarily affected in HUS. The patient usually manifests hemolytic anemia, thrombocytopenia, hypertension, and oliguric renal failure. Laboratory evaluation reveals a normal coagulation profile and hemoglobinuria. The pathologic process usually is confined to the kidney, although some patients have mild neurologic symptoms. Postpartum renal failure is probably the same entity, except that the pregnancy has already ended. Treatment in both cases consists of dialysis and red blood cell transfusions to maintain the hematocrit above 20%. Maternal morbidity and mortality are significant, with death frequently resulting from uncontrollable hemorrhage.
Coagulation Defects Von Willebrand Disease
Von Willebrand disease is an inherited defect of vWF, one of the proteins in the coagulation cascade. vWF is a large glycoprotein synthesized by endothelial cells and megakaryocytes and serves two functions: it is the plasma carrier for factor VIII, and it allows normal platelet aggregation at sites of endothelial injury. These two functions are directed by two different regions of the molecule, and several different mutations in both of these domains have been identified. There are three forms of von Willebrand disease.
Type I (approximately 75% of cases) and type II von Willebrand disease (25%) are inherited as autosomal dominant traits. Affected individuals have one normal vWF gene in addition to the abnormal gene, and some normal vWF will therefore be produced. As a result, individuals with type I disease are mildly affected, exhibiting easy bruising or bleeding only after dental procedures. Individuals with type II disease usually experience more severe bleeding problems, such as menorrhagia or corpus luteum hemorrhage. Type III disease is autosomal recessive and extremely
rare. It usually is associated with severe symptoms due to no normal production of the protein.
rare. It usually is associated with severe symptoms due to no normal production of the protein.
If the diagnosis is not made before pregnancy, it may be considered after excessive bleeding from a surgical or episiotomy site. Retrospectively, the patient may describe easy bruising or heavy menses. Pedigree analysis frequently includes similarly affected family members. The diagnosis is confirmed by all or some combination of the following laboratory tests: a prolonged bleeding time; decreased vWF concentration; reduced ristocetin cofactor activity; reduced factor VIII activity; and in some cases, mutational analysis.
Women with von Willebrand disease usually tolerate pregnancy well, in large part because the production of all coagulation factors is increased and vWF factor levels can approach near-normal levels. Despite this, the bleeding time may still be prolonged, and treatment may be required. If the bleeding time is prolonged at term, levels of vWF must be increased so that postpartum or surgical hemorrhage can be avoided. One way to increase the vWF level is to administer desmopressin acetate (DDAVP) for 48 hours prior to planned delivery. Patients with type I disease have the best response to desmopressin; those with type III disease usually do not respond at all. Thus, a trial of the therapy should be conducted in the second trimester. Alternatively, vWF replacement can be provided. Fresh frozen plasma contains all coagulation factors in equal proportions; cryoprecipitate contains factor VIII, vWF, and fibrinogen; and lyophilized factor VIII contains only that protein. For patients with von Willebrand disease, the recommended therapy is 15 to 20 U of cryoprecipitate given twice daily just prior to delivery and for 2 to 3 days afterward. Factor VIII concentrate can be administered instead. Effective treatment should normalize the bleeding time.
Women with type I or type II von Willebrand disease have a 50% risk of having an affected child; those with type III disease have minimal risk, unless they are related to their spouses. Prenatal diagnosis can be offered to at-risk pregnancies.
Hemophilias A and B
Hemophilias A and B are X-linked disorders of two genes that encode coagulation proteins. Hemophilia A results from mutations in a gene that is part of the factor VIII complex, while hemophilia B has mutations in the gene that encodes for factor IX. In female carriers, levels of these factors are thus reduced by one half or more. These decreased factor levels are adequate for normal hemostasis, and carrier women usually are clinically unaffected. In rare circumstances, a woman may exhibit all of the classic features of hemophilia (i.e., if she is homozygous for the mutation or if she is a carrier and has unfavorable lyonization). Such patients benefit from factor replacement.
Carrier females should be offered genetic counseling. Female offspring will be carriers, and one half of their sons will have hemophilia. Prenatal diagnosis is available. In ongoing affected pregnancies, knowledge that a male fetus carries a hemophilia gene allows the obstetrician to plan to avoid placing a scalp electrode during labor and to avoid vacuum-assisted or forceps-assisted vaginal delivery. Cesarean delivery should be for obstetric indications only, because atraumatic spontaneous vaginal delivery does not entail additional risk for the affected fetus.
Gastrointestinal Disease
Nausea and Vomiting
Mild and self-limited nausea and vomiting in the first trimester of pregnancy occur in 60% to 80% of women. Chronic nausea and vomiting, or hyperemesis gravidarum, complicates 1 in 200 to 300 pregnancies. This disorder is characterized by dehydration, electrolyte imbalance, and nutrition depletion and prompts medical intervention.
The etiology of hyperemesis is unclear. Theories have suggested the influence of human chorionic gonadotropin, the pituitary–adrenal axis, transient hyperthyroidism, psychogenic factors, and potential evolutionary benefit. Regardless of the cause, intervention is appropriate, ranging from intravenous hydration and antiemetic medications to nasogastric enteral feeding and hyperalimentation. Pregnancies complicated by mild or severe hyperemesis are not at increased risk for growth abnormalities, congenital anomalies, or prematurity (Table 17.4).
Gastrointestinal Reflux Disease
One half of all pregnant women complain of gastroesophageal reflux disease (GERD), commonly known as heartburn, sometime during pregnancy and particularly in the third trimester. Complaints include burning substernal discomfort with or without radiation, dysphagia exacerbated by meals, and increased intra-abdominal pressure, all worsening in the recumbent position. The differential diagnosis includes angina, achalasia, and structural or functional causes of dysphagia.
Risk factors for gestational GERD include heartburn prior to or in previous pregnancies, multiparity, and advanced gestational age. There is no association between GERD and race, prepregnancy weight, or weight gain during pregnancy. Treatment options are similar to treatment of the nonpregnant population, depend on the severity of symptoms, and are initiated sequentially beginning with lifestyle modifications and antacids. In severe refractory cases, cimetidine and metoclopramide are appropriate therapeutic interventions.
Peptic Ulcer Disease
Gastric secretion and motility are reduced and mucus secretion is increased during gestation. As a result, peptic ulcer disease (PUD) is uncommon in pregnancy, and its
complications, such as hemorrhage and perforation, are quite rare. Patients with PUD often experience considerable improvement, if not remission, of disease in pregnancy. However, PUD recurs in most women within 2 years of delivery.
complications, such as hemorrhage and perforation, are quite rare. Patients with PUD often experience considerable improvement, if not remission, of disease in pregnancy. However, PUD recurs in most women within 2 years of delivery.
TABLE 17.4 Gastrointestinal Disease in Pregnancy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Upper Gastrointestinal Bleeding
Hyperemesis can be accompanied by gastrointestinal bleeding. Although gastrointestinal bleeding prompts a concern for PUD with hemorrhage, most pregnant women with hematemesis will prove to have Mallory-Weiss tears. These small, linear mucosal tears near the gastroesophageal junction respond to iced saline lavage, antacids, and intravenous cimetidine. Endoscopy can be performed during pregnancy and will detect esophageal rupture with bleeding (Boerhaave syndrome), a much more serious diagnosis for which surgery and gastroenterology consultations are appropriate.
Cholelithiasis and Biliary Disease
Studies using serial ultrasonographic examinations over the course of pregnancy confirm that the risk of gallstones is increased, to an incidence of 2% to 10%, because pregnancy is characterized by decreased gallbladder motility and increased biliary sludge. Many women with cholelithiasis are relatively asymptomatic during pregnancy and require no intervention. However, acute cholecystitis complicates about 1 in 1,000 to 1,600 gestations. It is characterized by postprandial pain in the right upper quadrant or epigastric area, with radiation to the back or shoulder. This type of pain, with anorexia, nausea, emesis, low-grade fever, and leukocytosis, suggests stone obstruction of a duct. Ultrasonographic examination is very helpful, detecting approximately 95% of stones.
Management is the same as in a nonpregnant individual. Three fourths of patients with acute cholecystitis will respond to medical therapy consisting of bowel rest, nasogastric suction, intravenous hydration, antibiotics, and analgesics. The remainder will require surgical intervention for persistent pain, empyema, gangrene, or perforation. Open laparoscopic cholecystectomy during pregnancy is becoming more widely accepted. Although the second trimester is considered optimal for any surgical procedure, delay in treatment should be avoided regardless of gestational age.
Pancreatitis
Pancreatitis occurs with an incidence of 1 in 1,500 to 4,000 during pregnancy, with the majority of cases due to cholelithiasis. Other far less common etiologies include ethanol abuse, certain medications, trauma, and hypertriglyceridemia. Symptoms include midepigastric pain with back radiation, anorexia, nausea, and emesis. In normal pregnancy, serum amylase and lipase levels tend to increase only slightly with advancing gestation. The upper limits of normal for amylase and lipase in the first two trimesters are 100 U/dL and 200 U/dL, respectively. Significant elevations of these enzymes are therefore consistent with pancreatitis, although the degree of elevation does not correlate with disease severity. As in the nonpregnant population, pancreatitis is managed by bowel rest, nasogastric suction, analgesia, and intravenous hydration.
In most patients, inflammation subsides within 2 to 7 days. In the minority, abscess or pseudocyst formation prompts abdominal exploration. In this population, perinatal morbidity ranges from 5% to 15%, and perinatal mortality can be as high as 38%, most likely resulting from accompanying hypovolemia, hypoxia, and acidosis.
Inflammatory Bowel Disease
The term inflammatory bowel disease (IBD) refers to two forms of intestinal inflammation—namely, Crohn disease and ulcerative colitis. These diseases share many features but usually can be differentiated. IBDs have a significant genetic component and a fall under the classification of complex inheritance. The greatest risk factor for IBD is a family history of IBD. When both parents have IBD, the risk to the offspring is as high as 36% and is unaffected by disease activity in either parent at the time of conception. Figures for healthy offspring, congenital abnormalities, spontaneous abortions, and fetal demise are the same in pregnancies complicated by IBD as in the control population. Some report an increased risk of low birth weight in patients with Crohn disease, particularly if there is ileal disease, a history of bowel resection, or current tobacco abuse.
Ulcerative Colitis
Ulcerative colitis is a mucosal disease, almost always involves the rectum, and extends proximally and continuously for a variable distance. Symptoms include diarrhea, often with bleeding, and some degree of abdominal pain. Affected individuals also may have arthritis, uveitis, or erythema nodosum. Colon cancer, specifically adenocarcinoma, is increased in this population. The clinical course is one of exacerbations and remissions. The most serious complication is toxic megacolon, which can necessitate an emergency colectomy. Management is rapidly evolving, and new treatment regimens are based on the underlying mechanisms of disease and complement the older traditionally used drugs that include sulfasalazine, 5-aminosalicylic acid, and prednisone. If ulcerative colitis is quiescent at the time of conception, only one third to one half of patients will experience reactivation, often in the first trimester. Active disease at the time of conception may have a worse prognosis. When the disease is active, aggressive medical management, including parenteral nutrition, is essential.
Crohn Disease
Crohn disease is a transmural granulomatous inflammatory process that involves the rectum about 50% of the time. It may involve any part of the gastrointestinal tract but most often involves the terminal ileum and colon. “Skip” areas are common. Diarrhea and hematochezia can occur, and abdominal pain is almost always a problem. Nutritional deficiencies are more common than with ulcerative colitis. Complications include toxic megacolon and fistula formation, which is problematic for vaginal delivery if the perineum is involved. Eighteen percent of patients develop de novo perineal involvement after vaginal delivery, most often if an episiotomy was performed. As in ulcerative colitis, the patient also may have arthritis, and the risk of cancer is increased. Cancer risk correlates with the extent of mucosal pathology (pancolitis confers the highest risk) and the duration of the disease. In patients with long-standing disease, the risk exceeds 1% per year. Quiescent disease at conception carries a good prognosis. Similar to ulcerative colitis, there has been significant advances in the medical management of the disease. Surgery is necessary in about 5% of such pregnant patients.
Hepatitis
Acute viral hepatitis in pregnancy is a systemic illness with fever, nausea, emesis, and fatigue. Jaundice is common initially, and liver function tests are markedly elevated. With the exception of hepatitis E virus (HEV) infection, viral hepatitides do not occur more frequently or with greater severity in pregnancy. HEV infection is more dangerous in a pregnant patient, with a mortality of 15% to 20%. It is transmitted by the fecal–oral route and occurs most frequently in countries with poor sanitation (e.g., the Middle East, Africa, and India). Infection in the third trimester often is associated with fulminant hepatitis as well as preterm delivery and neonatal and maternal death.
Hepatitis A
Hepatitis A virus (HAV) is an RNA virus, with fecal–oral transmission and an incubation period of 15 to 50 days. This highly contagious disease is self-limited, with resolution over 2 to 3 weeks. Acute HAV infection is confirmed by a positive anti-HAV immunoglobulin M (IgM) antibody test. There are no chronic sequelae, and HAV does not cross the placenta. A single dose of hepatitis immune globulin is recommended as soon as possible after exposure. If the exposed pregnant patient becomes infected, close contacts, including the neonate, should be offered passive immunotherapy.
Hepatitis B
Hepatitis B virus (HBV) is a double-stranded DNA virus with worldwide distribution, transmitted by parenteral and sexual contact. Risk factors include multiple sexual partners, intravenous drug abuse, and receipt of blood products. Its incubation period is 40 to 100 days, and it can be recovered from all body fluids—most importantly, blood, breast milk, and amniotic fluid. HBV surface antigen (HBsAg) and anti-Hb core (anti-HBc) IgM antibody are seen in the early clinical phase of infection, before icteric changes or elevations in liver function tests. They indicate infectivity (Fig. 17.3). The presence of HBe antigen (HBeAg)
denotes active viral replication. Although HBeAg usually indicates acute infection, its persistence correlates both with the chronic carrier state and with the ultimate development of hepatocellular carcinoma. The risk of maternal–fetal transmission increases dramatically to 90% when acute infection occurs in the third trimester or in the presence of both HBsAg and HBeAg positivity and is a consequence of intrapartum exposure to blood and genital secretions. If the mother develops HBV infection remote from delivery and has developed anti-HB antibodies, the risk of fetal or neonatal infection is considerably less. The neonate’s risk of active or chronic disease is reduced significantly by HB immune globulin and the HBV vaccine; these should be given at delivery. Breast-feeding does not increase the risk of infection in these infants. The absence of HBsAg excludes active or chronic infection, and there is no risk for neonatal transmission. In the at-risk patient who is HBsAg negative and antibody negative, vaccination should be offered, as it is not contraindicated in pregnancy.
denotes active viral replication. Although HBeAg usually indicates acute infection, its persistence correlates both with the chronic carrier state and with the ultimate development of hepatocellular carcinoma. The risk of maternal–fetal transmission increases dramatically to 90% when acute infection occurs in the third trimester or in the presence of both HBsAg and HBeAg positivity and is a consequence of intrapartum exposure to blood and genital secretions. If the mother develops HBV infection remote from delivery and has developed anti-HB antibodies, the risk of fetal or neonatal infection is considerably less. The neonate’s risk of active or chronic disease is reduced significantly by HB immune globulin and the HBV vaccine; these should be given at delivery. Breast-feeding does not increase the risk of infection in these infants. The absence of HBsAg excludes active or chronic infection, and there is no risk for neonatal transmission. In the at-risk patient who is HBsAg negative and antibody negative, vaccination should be offered, as it is not contraindicated in pregnancy.
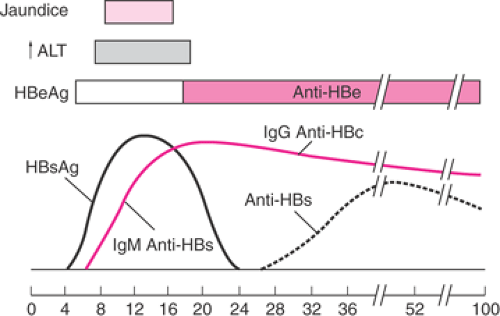 Figure 17.3 Timing of hepatitis B antigen and antibody production in acute hepatitis B infection. (ALT, alanine aminotransferase; HBeAg, hepatitis E antigen; HBsAg, hepatitis B surface antigen; IgG, immunoglobulin G; anti-HBc, antibody to hepatitis B core antigen; anti-HBs, antibody to hepatitis B surface antigen.) (From Dienstag JL, Isselbacher KJ. Acute hepatitis. In: Isselbacher KJ, Braunwald E, Wilson JD, et al., eds. Harrison’s principles of internal medicine, 13th ed. New York: McGraw-Hill, 1994:1458 , with permission.) |
Hepatitis C
Hepatitis C virus (HCV) is the agent primarily responsible for non-A, non-B (posttransfusion) hepatitis. HCV is a single-stranded RNA virus. Principal risk factors for HCV transmission are blood product transfusion and intravenous drug use. Acute HCV infection follows an incubation period of 3 to 60 days, and only 25% of infected patients will be symptomatic. The presence of HCV antibody indicates chronic infection and does not confer immunity; approximately one half of those infected develop chronic liver disease. No specific therapy has been shown to be efficacious in decreasing the morbidity of the disease. Coinfection with HCV and HIV is thought to accelerate the progression of hepatic injury.
Seroprevalence studies in pregnant patients in the United States indicate an incidence of HCV of 2% to 4%. Vertical transmission is proportional to the maternal HCV RNA titer, and approximately 8% of patients transmit the disease to their offspring. Coinfection with HIV is associated with an increased rate of perinatal transmission of 23% to 44%. Breast-feeding in the HCV-positive patient is not contraindicated by virtue of the 4% transmission rate in breast- and bottle-fed infants.
Hepatitis D
Hepatitis D virus (HDV) is an RNA virus that is dependent on coinfection with HBV for replication. HDV is acquired as a coinfection with HBV or as a superinfection in a chronic HBV carrier. Coinfection rarely leads to chronic disease, whereas superinfection is associated with an 80% likelihood of chronic hepatitis. Perinatal transmission of HDV can be prevented by the immunoprophylaxis used for HBV.
Pregnancy Following Liver Transplantation
Following liver transplantation, most authorities recommend that pregnancy be avoided for at least 12 months so that graft viability can be assessed and immunosuppression can be achieved and maintained with the lowest possible medication dosages. Thirty-eight percent of liver transplant patients are hypertensive; pregnancy does not increase this incidence or hasten graft rejection. The incidence of spontaneous abortion is similar to that of the general pregnant population, and the incidence of preeclampsia is 13.5%. Anemia complicates 31% of pregnancies in liver transplant patients, and rejection develops or worsens in 9%. Fifty-eight percent deliver at term, and the majority deliver appropriately grown babies vaginally. Consultation with a specialist in hepatology is recommended in pregnancy.
Acute Fatty Liver
Acute fatty liver of pregnancy (AFLP) has an incidence of 1 in 13,000 deliveries. AFLP accounts for a large percentage of severe liver disease in pregnancy and is accompanied by a mortality of up to 25%. Primiparity, male fetal sex, and multiple gestation appear to confer a higher risk. The etiology is unknown, and liver biopsy reveals microvesicular fatty infiltrates.
Symptoms typically appear in the late third trimester and include malaise, persistent nausea, and vomiting. Right upper quadrant or epigastric pain is noted in 50% to 80%. Laboratory abnormalities include elevated liver function tests, increased ammonia and uric acid levels, hemolysis, hypoglycemia, and coagulopathy. Early recognition is essential; if untreated, AFLP progresses to multiorgan system failure and death. Once it is diagnosed, intensive supportive care
is provided, and delivery is necessarily accomplished. Under these circumstances, maternal and fetal mortality are <20%. Survivors have no long-term sequelae, and recurrence in subsequent pregnancies is a rarity.
is provided, and delivery is necessarily accomplished. Under these circumstances, maternal and fetal mortality are <20%. Survivors have no long-term sequelae, and recurrence in subsequent pregnancies is a rarity.
Cardiovascular Disease
Physiologic Changes in Pregnancy
Normal pregnancy entails many physiologic changes that can stress the cardiovascular system. Plasma volume increases are measurable by 6 to 8 weeks gestation and 45% greater by 30 to 34 weeks. Red blood cell volume increases about 25%, resulting in a physiologic anemia. Cardiac output increases by 30% to 50% during the first half of pregnancy (as the result of an increase in both stroke volume and heart rate), by a further 30% during active labor, and by 45% during pushing. Systemic vascular resistance decreases during pregnancy, with both systolic and diastolic blood pressures falling during the second trimester and then returning to prepregnancy values in the third trimester. During labor, each uterine contraction results in an autotransfusion of 300 to 500 mL of blood. Cardiac output during this time is influenced by maternal vascular volume, maternal position, pain, and the method of pain relief (epidural anesthesia, spinal anesthesia, or intravenous narcotics). Cardiac output rapidly increases at delivery as the result of autotransfusion and relief of caval compression by the involuting uterus.
Women with cardiovascular disease may tolerate these physiologic changes poorly. Knowledge of the pregnancy-associated risks and complications associated with each type of heart disease allows the physician to choose management that optimizes the chances for a good pregnancy outcome. For each patient, the prepregnancy cardiovascular status should be established and used as a reference in assessing any pregnancy-related cardiac changes. The New York Heart Association (NYHA) classification scheme is useful for quantifying symptomatology:
Class I: patients are asymptomatic in all situations.
Class II: patients are symptomatic with greater-than-normal exertion.
Class III: patients are symptomatic with normal activities.
Class IV: patients are symptomatic at rest.
Although useful for categorizing symptoms, this classification scheme does not necessarily predict pregnancy outcome. In one large retrospective study, for example, the majority of cases of pulmonary edema and maternal death occurred in women who were functional class I or class II. However, this scheme can be used to assess changes in cardiac function. Any change in cardiac classification during the pregnancy, even if only from class I to class II, can be ominous and should prompt a thorough evaluation and aggressive management. Bed rest or hospitalization often is required.
Rheumatic Heart Disease
Approximately 4% of reproductive-age women have heart disease. Although this number has remained fairly constant, the relative incidence of the various forms of heart disease has changed dramatically during the last few decades. During most of the 20th century, the majority of heart disease resulted from rheumatic fever (group A β-hemolytic Streptococcus); the ratio of rheumatic heart disease to congenital heart disease was 20 to 1. During the last few decades, however, the prevalence of rheumatic heart disease has decreased significantly, while the number of adult survivors with congenital heart disease has increased; the ratio is now 3 to 1 or less. Nevertheless, rheumatic valvular disorders still account for a substantial proportion of heart disease in reproductive-age women.
Mitral Stenosis
Mitral stenosis is the most common form of rheumatic heart disease in women. Rheumatic fever typically occurs between ages 6 to 15 years. If myocarditis is present, mitral insufficiency will develop, followed in approximately 5 years by mitral stenosis. Symptoms usually do not begin for another 15 years after that, with severe complications such as right-sided heart failure occurring in another 5 to 10 years. The mean age for the initiation of symptoms is thus 31, with incapacity occurring at age 38 if the condition is not treated. Initial symptoms include fatigue and dyspnea on exertion, which progress to dyspnea at rest and hemoptysis. Atrial arrhythmias, infection, or pulmonary embolism can lead to heart failure.
The stenotic mitral valve impairs left ventricular filling and thus limits any increase in cardiac output. Pregnancy-mediated cardiovascular changes, especially increased intravascular volume and increased heart rate, can exacerbate the impaired filling and lead to decompensation during pregnancy and especially during labor, delivery, and the puerperium. Left atrial volume and pressure increase, pulmonary venous pressure increases, and, eventually, features of pulmonary hypertension and right ventricular hypertrophy and failure can develop. The goals of management are to optimize cardiac output by preventing rapid ventricular rates and avoiding decreases in systemic vascular resistance, to reduce stress on the right ventricle by minimizing increases in blood volume, and avoiding situations in which pulmonary artery pressure is increased (i.e., hypercarbia, hypoxia, or acidosis). Two serious complications associated with mitral stenosis are atrial fibrillation and pulmonary edema. Both have been associated with maternal death.
Tachyarrhythmias that occur in the patient during pregnancy should be treated, because a rapid heart rate prevents adequate ventricular filling and decreases cardiac output.
β-Blockers should be considered for the patient with a heart rate above 90 beats per minute. Digoxin and heparin may be required for the patient with atrial fibrillation. Rarely, surgery becomes necessary during the pregnancy, including balloon valvuloplasty and surgical commissurotomy. During labor, bedside cardiac monitoring is routine; central hemodynamic monitoring is routine if the patient is in NYHA class III to IV or the valve diameter is <2.5 cm2. Pain must be managed effectively. Epidural anesthesia can be used if care is taken not to overload the patient with fluid beforehand and not to decrease systemic vascular resistance during the infusion. Fluid management must be meticulous, with extra attention given to the patient during the immediate postpartum period, when autotransfusion rapidly increases the central blood volume. Pulmonary function must be followed closely for pulmonary edema. A pulmonary artery catheter may assist in the management of patients with severe disease. Because the pulmonary capillary wedge pressure (PCWP) may not accurately reflect left ventricular filling pressure in severe mitral stenosis, the PCWP should be maintained in the high-normal to elevated range. If general anesthesia becomes necessary, agents that produce tachycardia (e.g., atropine, meperidine, ketamine) should be avoided. The high-risk period for severe decompensation continues for 24 to 48 hours postpartum.
β-Blockers should be considered for the patient with a heart rate above 90 beats per minute. Digoxin and heparin may be required for the patient with atrial fibrillation. Rarely, surgery becomes necessary during the pregnancy, including balloon valvuloplasty and surgical commissurotomy. During labor, bedside cardiac monitoring is routine; central hemodynamic monitoring is routine if the patient is in NYHA class III to IV or the valve diameter is <2.5 cm2. Pain must be managed effectively. Epidural anesthesia can be used if care is taken not to overload the patient with fluid beforehand and not to decrease systemic vascular resistance during the infusion. Fluid management must be meticulous, with extra attention given to the patient during the immediate postpartum period, when autotransfusion rapidly increases the central blood volume. Pulmonary function must be followed closely for pulmonary edema. A pulmonary artery catheter may assist in the management of patients with severe disease. Because the pulmonary capillary wedge pressure (PCWP) may not accurately reflect left ventricular filling pressure in severe mitral stenosis, the PCWP should be maintained in the high-normal to elevated range. If general anesthesia becomes necessary, agents that produce tachycardia (e.g., atropine, meperidine, ketamine) should be avoided. The high-risk period for severe decompensation continues for 24 to 48 hours postpartum.
Although the American Heart Association recommends antibiotic prophylaxis only for women who have a vaginal delivery in the presence of an infection or who undergo urethral catheterization, many clinicians provide prophylaxis to all cardiac patients. Subacute bacterial endocarditis (SBE) prophylaxis usually includes ampicillin 2 g and gentamicin 1.5 mg/kg intravenously 30 minutes before delivery and ampicillin 1 g intravenously or amoxicillin 1 g orally 6 hours after delivery. Penicillin-allergic patients should receive vancomycin.
Mitral Insufficiency
Mitral insufficiency results in regurgitation of blood from the left ventricle back into the left atrium, with resulting left atrial enlargement. Most patients tolerate mitral insufficiency well and remain asymptomatic for 30 to 40 years. However, because pulmonary edema or embolism, atrial tachycardia, and infective endocarditis can occur during pregnancy, patients with mitral insufficiency should be monitored closely. Anything that stresses or impairs the function of the left ventricle should be avoided. Increases in systemic vascular resistance, atrial fibrillation, bradycardia, or myocardial depressants can all result in left ventricular decompensation. During labor, pain should be treated effectively and fluid management calculated to maintain left ventricular volume without increasing it. Epidural anesthesia can be very effective, as long as preprocedure hydration is conducted cautiously. SBE prophylaxis should be given. Occasionally, surgical valve replacement is necessary during pregnancy.
Aortic Insufficiency
Aortic insufficiency (AI) usually occurs 7 to 10 years after an episode of rheumatic fever myocarditis, and the patient remains asymptomatic for another 7 to 10 years. The regurgitant valve causes a chronic increase in left ventricle volume, eventually leading to increased compliance, increased end-diastolic pressure, and pulmonary congestion and edema. Most pregnant women with AI are relatively asymptomatic. This is, in part, because the decreased systemic vascular resistance and increased heart rate typical of pregnancy tend to increase forward flow through the insufficient valve. However, cardiovascular changes occurring during labor and delivery can lead to decompensation, especially if intravascular volume is increased markedly or systemic vascular resistance is increased by pain or other stressors.
Epidural anesthesia is ideal for such patients, as it eliminates pain and decreases systemic vascular resistance. However, care must be taken not to reduce diastolic blood pressure or provoke a bradycardic episode, because left ventricular output will decrease as a result. Myocardial depressants should be avoided, and fluids must be managed carefully to maintain adequate volume but not overload the left side of the heart. Frequent pulmonary examinations to rule out pulmonary congestion may be helpful. SBE prophylaxis should be given.
Aortic Stenosis
Aortic stenosis (AS) resulting from rheumatic fever rarely complicates pregnancy, because the time lag between the rheumatic fever episode and the occurrence of stenosis is usually 35 to 40 years. However, AS can occur in reproductive-age women, and those who are symptomatic (e.g., angina, syncope, shortness of breath) have a risk of sudden death out of proportion to the severity of their symptoms; left ventricular failure and infective endocarditis are other serious complications.
The normal cross-sectional area of the aortic valve is 2.6 to 3.5 cm2; an orifice <2.6 cm2 usually is heralded by a loud systolic murmur, while an orifice <1 cm2 produces symptoms of dyspnea, chest pain, and syncope. AS results in a relatively fixed stroke volume that is dependent on both adequate diastolic filling and heart rate. Although some increase in heart rate helps to maintain an adequate cardiac output, tachycardia >140 beats per minute, bradycardia, and decreased systemic vascular resistance are poorly tolerated.
For these reasons, use of epidural anesthesia is controversial for pain relief during labor, and the patient could instead be managed with parenteral narcotics and pudendal block. Fluid management must be meticulous, taking care to maintain an adequate intravascular and thus end-diastolic volume. A pulmonary artery catheter may be quite helpful in directing fluid management. Because hypovolemia is a far greater threat to this patient than is pulmonary edema, the pulmonary artery wedge pressure
should be maintained in the range of 14 to 16 mm Hg to provide a margin of safety against unexpected peripartum blood loss.
should be maintained in the range of 14 to 16 mm Hg to provide a margin of safety against unexpected peripartum blood loss.
Congenital Heart Disease
Congenital heart disease accounts for the majority of all heart disease in reproductive-age women. Many women now reach adulthood without surgical correction of their lesions, while for others, early surgery has been lifesaving. Women who have undergone surgical correction, have normal hemodynamics, and are completely asymptomatic generally tolerate pregnancy, labor, and delivery well without special considerations. Women with uncorrected lesions, however, require special management. The most common uncorrected heart abnormalities seen in pregnancy are atrial septal defect (ASD), patent ductus arteriosus (PDA), ventricular septal defect (VSD), pulmonic stenosis, congenital AS, coarctation of the aorta, and tetralogy of Fallot.
Both maternal and fetal outcomes depend on the nature of the cardiac lesion, the patient’s functional capacity, the history of surgical repair (if any), and the presence or absence of pulmonary hypertension or cyanosis. In the presence of cyanosis, there is an increased risk of functional deterioration, congestive heart failure, maternal mortality, IUGR, preterm birth, miscarriage, and stillbirth. In one series, only 55% of pregnancies in cyanotic mothers resulted in a live birth.
A woman with congenital heart disease should receive genetic counseling regarding the etiology of the lesion and risks to her fetus. Isolated congenital heart malformations are considered multifactorial in origin and thus have a general recurrence risk of 3% to 10% in first-degree relatives. However, a more precise recurrence risk can be provided if the heart defect is categorized according to the aspect of abnormal cardiac development. Many structural cardiac defects can be identified by second-trimester ultrasonographic examination or fetal echocardiogram.
Mitral Valve Prolapse
Mitral valve prolapse (MVP) is the most common congenital valvular lesion, with an incidence of 5% to 10% in the general population. The majority of patients with MVP are asymptomatic and tolerate pregnancy, labor, and delivery well. Arrhythmias occur occasionally. Although the patient’s cardiovascular status should be monitored closely, usually no special therapy is required other than SBE prophylaxis, although this is controversial and should be based on the recommendations of a cardiologist.
Left-to-Right Intracardiac Shunts
Left-to-right intracardiac shunts can result from ASDs, VSDs, or PDAs. Small shunts often are well tolerated for many years. If there is no pulmonary hypertension and the patient is asymptomatic, pregnancy does not impose significant increased risk and may actually improve cardiac hemodynamics, as the decreased systemic vascular resistance encourages forward flow. Increased systemic vascular resistance or increased maternal heart rate may increase the shunt and should be avoided; epidural anesthesia for labor and delivery can be helpful. Patients with ASDs are at increased risk of developing supraventricular dysrhythmias that should be controlled with medication.
If, however, the shunt is substantial, resulting in many years of increased pulmonary blood flow, pulmonary hypertension and right heart failure can develop, and the shunt reverses. The combination of pulmonary hypertension and right-to-left shunt through any communication between the systemic and pulmonary circulation is known as Eisenmenger syndrome. This condition is life threatening in the pregnant patient, with a maternal mortality of 40% to 60%. Death is due to congestive heart failure and thromboembolic phenomena. The outcome for the fetus also is exceptionally poor, with a perinatal mortality exceeding 28% and a 55% incidence of preterm birth. Women with Eisenmenger syndrome should be strongly discouraged from becoming pregnant or carrying a pregnancy. Management of the gravid patient with this condition includes hospitalization, oxygen therapy, prophylactic anticoagulation, and treatment of heart failure with digoxin and diuretics. Delivery usually requires pulmonary artery catheterization, intrathecal morphine provides excellent analgesia without significant motor or autonomic effects, and shortening of the second stage of labor with forceps delivery is common. SBE prophylaxis is routine, and many consider minidose heparinization postpartum.
Tetralogy of Fallot
Right-to-left shunting is seen also in tetralogy of Fallot, which describes the association of VSD, right ventricular outflow tract obstruction, right ventricular hypertrophy, and overriding aorta. The amount of right-to-left shunting is determined by both the size of the VSD and the degree of right ventricular outflow tract obstruction. Uncorrected tetralogy of Fallot is a cyanotic condition characterized by decreased arterial oxygen saturation and polycythemia. Pregnancy can cause further decompensation, because the decreased systemic vascular resistance increases the right-to-left shunt; shunting is increased as well by a rise in the pulmonary vascular resistance resulting from the stress of labor. With uncorrected tetralogy of Fallot, 40% of women develop heart failure during pregnancy, and 12% die; the fetal mortality rate is high. Pregnancy is discouraged in those with uncorrected tetralogy. Poor prognosis is associated with several factors, including a prepregnancy hematocrit of over 65%, a history of syncope or congestive heart failure, electrocardiographic evidence of right ventricular strain, and a peripheral oxygen saturation of <80%. Pregnancy management includes bed rest, oxygen therapy, and isotopic support as necessary. Because any decrease in systemic vascular resistance can be life threatening, epidural
or spinal anesthesia should be avoided. Intravenous medication and pudendal block can be used, and the second stage of labor should be shortened.
or spinal anesthesia should be avoided. Intravenous medication and pudendal block can be used, and the second stage of labor should be shortened.
Congenital Aortic Stenosis
Congenital AS accounts for 5% of all congenital heart disease, with bicuspid aortic valve being the most common malformation. Many patients with bicuspid aortic valve are completely asymptomatic and tolerate pregnancy, labor, and delivery well. For those who are symptomatic, management considerations are the same as for AS resulting from rheumatic heart disease. Bicuspid aortic valves can be a heritable trait.
Coarctation of the Aorta
Coarctation of the aorta rarely complicates pregnancy, because most affected women undergo surgical correction as children. During pregnancy, patients with uncorrected coarctation face an increased risk of aortic dissection and rupture, and thus an increased risk of maternal (up to 9%) and fetal (20%) death, as well as bacterial endocarditis and cerebral hemorrhage (associated with intracranial aneurysms). Because the coarctation results in a fixed stroke volume, management is similar to that for AS.



