Neonatal Hyperammonemia
INTRODUCTION
The most common concern for neonates who present with poor feeding, tachypnea, hypothermia, and lethargy during the first days of life is sepsis; in fact, this is the most likely cause of severe illness. However, it is important to also consider the possibility of an inborn error of metabolism. Although individually these are rare disorders, as a group they probably are present in more than 1 in 5000 live births1; if they are not identified early, most die or are left with severe brain damage.
Many inborn errors that present in the newborn period with a catastrophic illness have elevated ammonia levels as a presenting symptom. It is our recommendation that every neonate who is worked up for sepsis should also have a plasma ammonia level drawn. While expanded newborn screening has been helpful in diagnosing a number of causes of neonatal hyperammonia, the results are often not available until the outcome of the disease is unalterable. At best, they become available when the child is showing symptoms and may aid in making the diagnosis. In this chapter, we discuss the clinical presentation, laboratory evaluation, and clinical effects of neonatal hyperammonemia. Chapter 106 provides a clinical management template for intervention in neonatal hyperammonemia.
DEFINING HYPERAMMONEMIA AND OBTAINING A SPECIMEN
Typical plasma ammonia levels in neonates are less than 40 μmol/L, similar to that in older children and adults. Premature infants may have somewhat higher levels (in the range of 40–60 μmol/L) because of the immaturity of their liver function and lower production of arginine during the first few weeks of life.2 Clinical symptoms attributable to hyperammonemia in the newborn generally occur above levels of 150 μmol/L, and it is not uncommon for neonates with urea cycle disorders to have plasma ammonia levels in the 1000–2000 μmol/L range.
The measurement of plasma ammonia can be affected by the collection technique, so a free-flowing sample is recommended. Plasma, once obtained, should immediately be placed on ice, taken to the laboratory, and analyzed within 30 minutes. With delayed measurement after sample collection, glutamine is deaminated to glutamate, which can lead to false elevation of plasma ammonia. Levels of plasma ammonia over 200 μmol/L in the neonate are highly suggestive of an underlying inborn error of metabolism and require immediate intervention.
CLINICAL PRESENTATION OF HYPERAMMONEMIA IN THE NEONATE
The classic presentation of neonatal hyperammonemia is as a catastrophic illness in the first week of life. Signs and symptoms of hyperammonia in the neonate depend on the proximate cause and the rapidity of ammonia accumulation. In patients with rapidly accumulating ammonia, presentation may occur in the first 2–3 days of life with lethargy and poor feeding. This may progress over a matter of hours to temperature instability, coma, and evidence of increased intracranial pressure. With more indolent ammonia accumulation, initial presentation may be a poor suck, vomiting, intermittent encephalopathy, and seizures. The symptoms of hyperammonemia may be cryptic and nonspecific early in the course, so assessment of plasma ammonia is critical in any neonate with systemic symptoms without a known or obvious cause.3–5
The electroencephalographic (EEG) pattern during hyperammonemic coma is one of low voltage with slow waves and asymmetric delta and theta waves. It may show a burst suppression pattern, and the duration of the interburst interval may correlate with the height of ammonium levels.6 Neuroimaging studies show cerebral edema with small ventricles, flattening of cerebral gyri, and diffuse low density of white matter; there may also be intracranial hemorrhage.7
DIFFERENTIAL DIAGNOSIS OF NEONATAL HYPERAMMONEMIA
Hyperammonemia is the result of either primary or secondary failure of nitrogen clearance. Ammonia is generated by the breakdown of amino acids and is converted into urea via the urea cycle (Figure 59-1). Hyperammonemia can result from primary failure of the urea cycle (urea cycle disorders); secondary impairment of the urea cycle by other inborn errors of metabolism (eg, organic acidemias, fatty acid oxidation [FAO] defects, mitochondrial disorders); generalized liver failure; or mechanical or clinical disease that increases protein breakdown that overwhelms the urea cycle (eg, port-systemic shunt, sepsis, crush injuries, and status epilepticus).
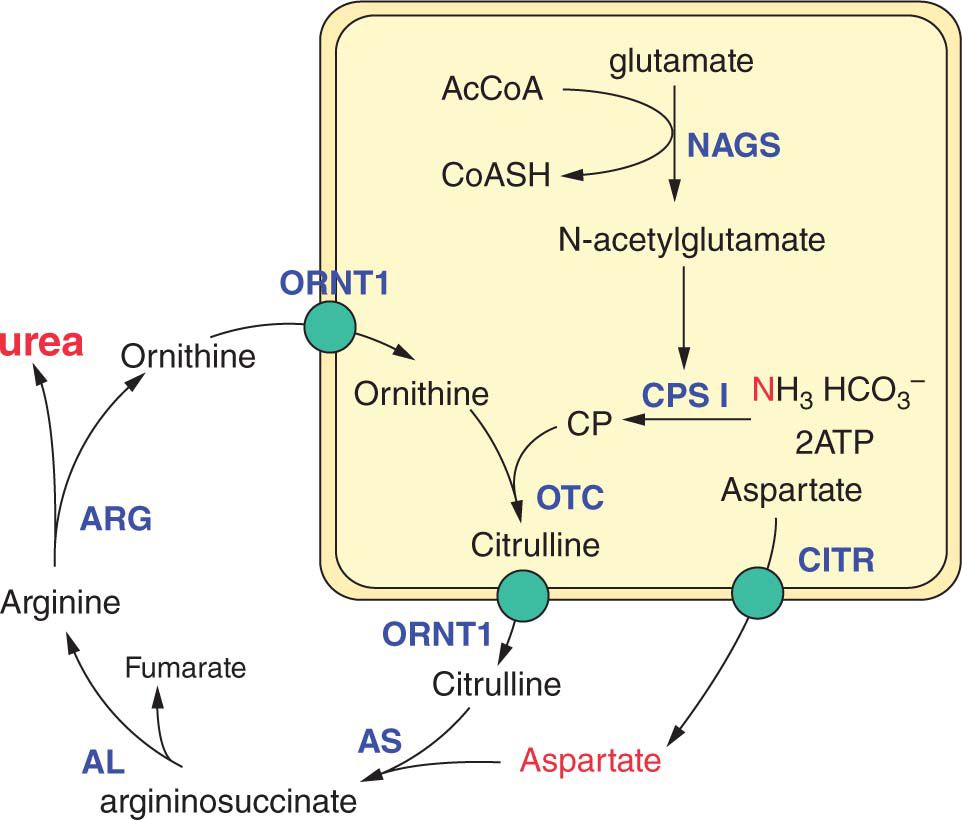
FIGURE 59-1 The urea cycle. AcCoA, acetyl-coenzyme A; AL, argininosuccinate lyase; Arg, arginase; AS, argininosuccinate synthetase; CITR, citrin; CoASh, coenzyme A; CPSI, carbamylphosphate synthetase I; NAGS, N-acetylglutamate synthetase; ORN1, ornithine translocase; OTC, ornithine transcarbamylase.
In the newborn period, hyperammonemia due to an inborn error of metabolism has a similar presentation to a number of acquired conditions, including sepsis, intracranial hemorrhage, congenital heart disease, and acute gastrointestinal events such as intussusception or volvulus. As noted, the measurement of plasma ammonia levels is critical for distinguishing between these conditions. The acquired disorders generally do not have significant elevations in ammonium levels, but the inborn errors of metabolism do. On the other hand, elevated lactate levels may be seen at initial presentation in all of these conditions other than urea cycle disorders.
A number of inborn errors of metabolism can cause hyperammonemia; these include urea cycle disorders, organic acidemias, nonketotic hyperglycinemia, congenital lactic acidoses, lysinuric protein intolerance, and FAO disorders. Lysinuric protein intolerance, however, rarely presents in the newborn period, and only rare severe long-chain FAO disorders present with hyperammonemia in the newborn.
Once hyperammonemia is identified, widely available tests with rapid turnaround times help narrow the differential diagnosis and guide initial intervention. First-line tests that should be collected after hyperammonemia is identified include a comprehensive metabolic panel, urinalysis, and arterial blood gas. If coagulopathy and elevated transaminases are present, then generalized hepatic failure is likely the cause. If not, then a specific inherited metabolic disease or structural anomaly leading to hyperammonemia must be considered.
Often, the first available laboratory data are the blood gas results. Respiratory alkalosis in the setting of hyperammonemia is specific and typical for primary urea cycle disorders. Acidosis would be consistent with an organic academia, congenital lactic acidosis, sepsis, or other causes of hyperammonemia. An elevated anion gap is typical in organic acidemias. Urinalysis for ketone measurement is fast and useful as ketones are unusual in neonates, even when critically ill. Significant ketone accumulation is suggestive of an organic acidemia. Standard electrolyte measurement can provide information about an elevated anion gap (which would suggest an organic acidemia or lactic acidosis). A low or absent serum urea nitrogen (BUN) may be seen with impaired urea cycle function. Liver function tests (coagulation profiles and transaminases) may reveal global hepatic dysfunction.8
Additional laboratory tests to be collected immediately include plasma amino acid (PAA) analysis, urine organic acid analysis, and plasma acylcarnitine profile. The results for these tests will not likely be available until after intervention has begun, but they will help identify the underlying cause for the hyperammonemia and guide more specific treatment.
Other clinical signs and symptoms may help narrow the differential diagnosis. In the organic acidemias (eg, propionic acidemia, methylmalonic acidemia, isovaleric acidemia, glutaric acidemia type II, and multiple carboxylase deficiency), hypoglycemia and pancytopenia may also be present. Mass spectrometric analysis of urine organic acids or blood acylcarnitine profile will yield the specific diagnosis. In these disorders, PAA analysis (but not cerebrospinal fluid [CSF]) may show an elevated glycine level, from which the initial name, ketotic hyperglycinemia, was derived. This is contrasted with nonketotic hyperglycinemia, in which there is the absence of ketosis, elevated glycine levels in both plasma and CSF, and an increased CSF/plasma glycine ratio.9
Fatty acid oxidation defects are caused by different chain length acyl-coenzyme A dehydrogenase deficiencies. Patients with these disorders typically present with hypoglycemia and no or little ketosis. There may be signs of liver disease and often heart involvement. Patients with severe long-chain FAO disorders occasionally present with hyperammonemia in the newborn period. Urinary organic acids show a dicarboxylic aciduria; plasma carnitine levels may be low, and abnormal acylcarnitine species are found.10
Congenital lactic acidosis can be the result of a primary deficiency of pyruvate carboxylase or pyruvate dehydrogenase or can be secondary to a defect within the mitochondrial respiratory chain. The principal biochemical finding is lactic acidosis. In pyruvate dehydrogenase deficiency and mild pyruvate carboxylase deficiency, the ratio of lactate to pyruvate is maintained at 10–20:1. In severe pyruvate carboxylase deficiency, mitochondrial respiratory chain defects, and secondary lactic acidosis, however, this ratio is significantly increased.11 In these oxidative phosphorylation defects and in other mitochondrial diseases, the 3-hydroxybutyrate/acetoacetate ratio is also elevated.
Patients with primary urea cycle disorders show a normal urinary organic acid profile, and the plasma lactate level is generally normal or mildly increased. Although peak plasma ammonium levels are typically in the range of 100–500 μmol/L in organic acidemia and congenital lactic acidosis, in neonatal onset urea cycle disorders they are generally above 500 μmol/L.
Plasma amino acid patterns are also distinct in urea cycle disorders (Figure 59-2), with low levels of arginine and abnormal (low or high) levels of citrulline. Citrulline is the product of carbamyl phosphate synthetase (CPSI) and ornithine transcarbamylase (OTC) and the substrate for argininosuccinate synthetase (AS) and argininosuccinate lyase (AL). Thus, its level is absent in N-acetyl-glutamate synthetase (NAGS), CPSI, and OTC deficiencies, and it is markedly elevated in AS and AL deficiencies.12 Differentiation of AS from AL deficiencies depends on finding argininosuccinic acid (ASA) in plasma and urine in AL deficiency. In ornithine translocase deficiency (HHH syndrome), there is accumulation of homocitrulline in the urine, and in argininemia, very high arginine levels are observed. HHH syndrome, however, has not been described as causing neonatal illness, and argininemia rarely presents in the neonatal period.13
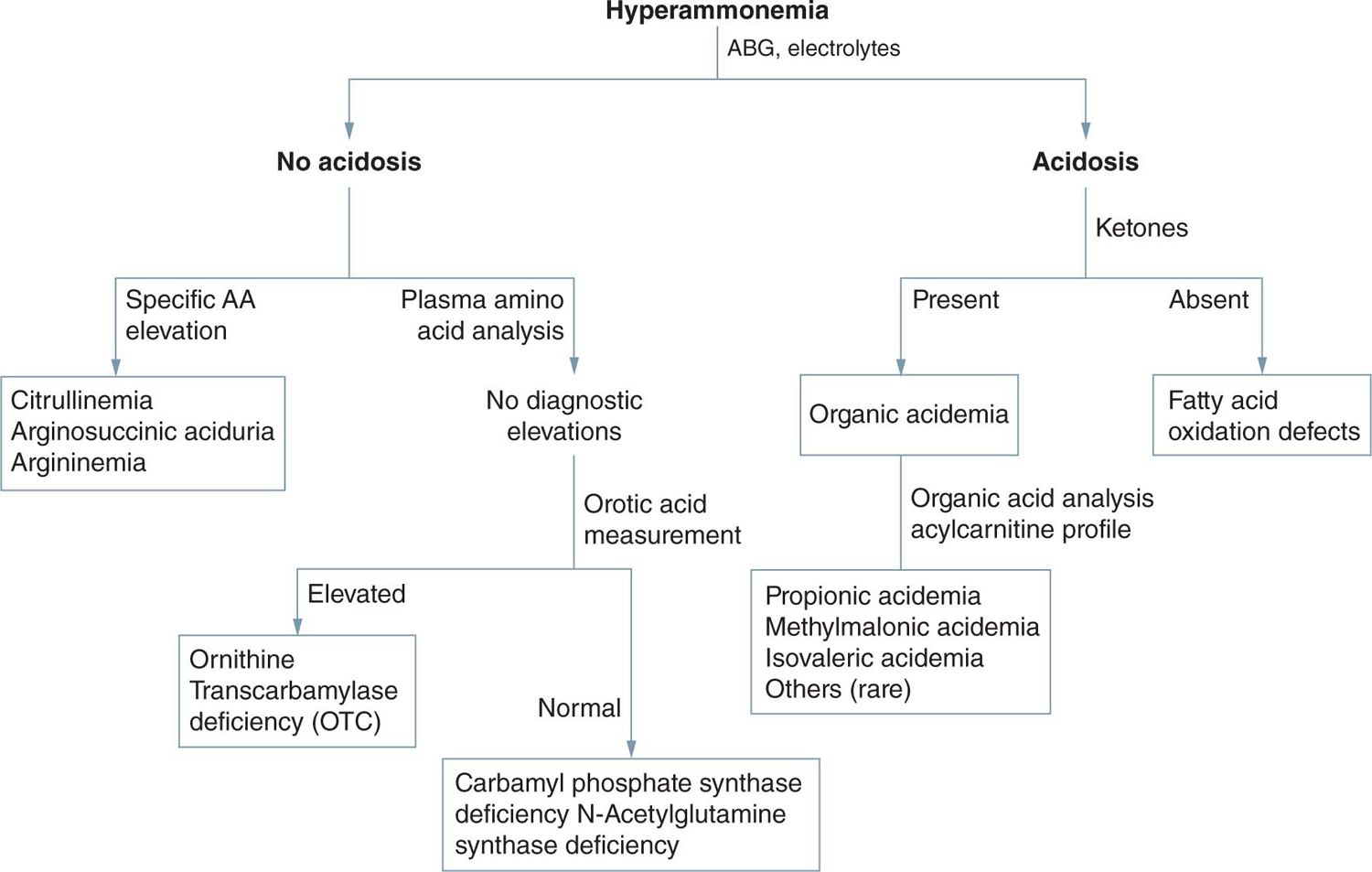
FIGURE 59-2 Urea cycle disorders. AA, amino acid; ABG, arterial blood gas.
Distinction of CPSI from OTC deficiency is dependent on detecting excessive urinary orotic acid excretion in OTC deficiency and normal or decreased excretion in CPSI deficiency. A deficiency of NAGS is biochemically similar to CPSI deficiency, with low or normal orotic acid excretion.14
In summary, it is likely that measurement of ammonium, amino acids, and lactate in blood and of amino acids, organic acids, and orotic acid in urine will identify almost all genetic causes of neonatal hyperammonemia. Confirmation of the diagnosis may require enzymatic or mutation analyses but is not necessary prior to beginning treatment.
TREATMENT OF NEONATAL HYPERAMMONEMIC COMA
The treatment of neonatal hyperammonemic coma requires a highly coordinated team of specialists.15,16 Emergency management is based on 3 interdependent principles: (1) physical removal of the ammonia by dialysis; (2) reversal of the catabolic state through caloric supplementation, aided by insulin as needed to maintain high-set-point normoglycemia; and (3) reduction of nitrogen intake as well as pharmacologic scavenging of waste nitrogen. These are not to be implemented consecutively but should be pursued independently and in parallel as rapidly as possible.8–10 (Table 59-1). A detailed approach to the treatment of hyperammonemia can be found in chapter 106.
Table 59-1 Treatment and Organization



