Fig. 5.1
Facial postinflammatory hypopigmentation due to prior atopic dermatitis lesions
Table 5.1 demonstrates a broad overview of loss of pigmentation in children, reviewing patterns of information and techniques to enhance diagnostic accuracy [1–8].
Table 5.1
Diagnosis of hypopigmentation reviewed in this chapter
Type of hypopigmentation | History | Wood’s lamp | Morphology/distribution | Dermoscopy | Biopsy | Further testing |
|---|---|---|---|---|---|---|
Pigmentary mosaicism | Birth to 2 years | Equivocal | Linear in the lines of Blaschko; Multiple Lesions can signal hypomelanosis of Ito or genetic abnormalities | NC | Only for fibroblast culture and genetic testing | Screening for other systems affected, e.g., neurological alterations can aid in long-term care |
Incontinentia pigmenti, 4th stage | Few weeks of life to mid-childhood; follows prior vesiculation, verrucous changes, or hyperpigmentation in the same distribution | Negative | Linear lesions in the lines of Blaschko | NC | Would be beneficial only in the vesicular or verrucous stage | Genetic testing and evaluation of family member |
Menkes kinky hair carrier | Congenital through adulthood | Negative | Linear along broad lines of Blaschko | NC | NC | Genetic testing and evaluation of family member |
Nevus depigmentosus | Birth through age 2 years | Negative | Annular area of partial pigment loss | NC | Genetic alterations could be detectable through culture of cells in this area and normal areas of pigmentation | Mexameter can differentiate from vitiligo Not necessary unless morphology is more consistent with ash leaf macules of tuberous sclerosis |
Morphea | Childhood or adolescent acute onset; pruritus, burning, or pain can be associated; linear lesions (segmental form) can be noted and are associated with limb movement limitation and intracranial problems | Positive in some cases | Annular violaceous bound down plaques over the trunk or extremities | Vascular ectasia, atrophic epidermis might be noted | Biopsy would confirm morphea diagnosis | Not necessary unless linear lesions are noted over the head or neck |
Lichen striatus | Onset in Spring, Summer, or fall abrupt and progressive distally to proximally along the extremities or on the face | Negative | Linear grouped small erythematous to flesh-colored papules resolving with hypopigmentation along the lines of Blaschko | Shows vascular ectasia and hyperkeratosis | Biopsy can demonstrate ball and claw lymphocytic clusters in the dermis | None needed |
Postinflammatory pigmentary alteration | Any age, following the occurrence of cutaneous inflammation | Negative | Morphology consistent with previous insult | NC | Biopsy demonstrates presence of melanocytes | None needed |
Waardenburg’s syndrome | Congenital onset | NC | Hypopigmentation of the anterior shins with islands of repigmentation; typical clinical appearance, e.g., heterochromic irides, dystopia canthorum | NC | Biopsy may demonstrate absence of melanocytes | Genetic testing/counseling; Hearing screen; Check for signs of Hirschsprung’s disease (rectal exam) |
Piebaldism | Congenital onset | NC | White forelock Irregular white patches on face, trunk, and extremities often symmetrically distributed | NC | Biopsy may demonstrate absence of melanocytes | Genetic testing, family history |
Mycosis fungoides | School-aged to adolescents | Positive in hypopigmented variant | Trunk, buttocks, and proximal extremities symmetric hypopigmented plaques with fine hyperkeratosis | Fine hyperkeratosis; vascular ectasia mild | Atypical lymphocytes; T-cell gene rearrangements | Good long-term prognosis |
Nutritional Deficiencies – Kwashiorkor – Fad diets/food avoidance – Eating disorders – Gastrointestinal disease (IBD, pancreatic insufficiency, gastric bypass surgery) – Vitamin B12 deficiency – Vitamin D deficiency – Selenium deficiency – Homocysteinuria | Any aged child | Negative | Generalized hypopigmentation; hyperkeratosis; central enhancement | NC | May depend on the deficiency; often with pallor and ballooning of upper epidermis | Associated with malnutrition and gastric bypass procedures in adolescents. Type of cutaneous alteration depends on specific nutritional deficiency |
Vitiligo | Onset in Spring of progressive pigment loss | Positive for highlighting | Hypopigmentation with good demarcation anywhere on the body, especially periorificial and intertriginous or over the joints of the extremities | NC | Biopsy would confirm the absence of melanocytes and potentially demonstrate lymphocytic infiltration at the active border | Mexameter can differentiate from nevus depigmentosus Thyroid tests and vitamin D levels are monitored standardly, but screening for other autoimmune illnesses can be performed on a case-by-case level |
Oculocutaneous albinism | At birth | Negative | Generalized Hypopigmentation with or without pigmentary loss of the eye and/or hair | NC | Biopsy would confirm presence of melanocytes | Genetic testing and family history/screening may reveal the type associated with pigment loss |
Tuberous sclerosis | At birth | Negative or equivocal | Hypopigmentation in format of thumbprints, confetti-like macules, and/or ash leaf macules; Later in childhood adenoma sebaceum, shagreen patch, and/or Koenen’s tumor | NC | Biopsy pattern is in the differential of normal; however less melanin would be noted with special stains if compared to normal skin | Annual skin examination. Genetic screen, MRI of the head, neurologic evaluation, cardiac evaluation, nephrology evaluation, and developmental evaluation may all be appropriate in these patients. |
Tey recently described a basic paradigm to help diagnose hypopigmentation of childhood [6]. The classification developed is based on age at diagnosis, with division as “early” onset by 2 years of age vs. “late” onset age two or older. Further division by localized vs. generalized illness helps to discern diagnosis. The paradigm has one caveat: many congenital lesions have late expression, particularly in light-skinned children (e.g., nevus depigmentosus and pigmentary mosaicism). Furthermore there are rare situations where autoimmune or autoinflammatory diseases are congenital or extremely early in onset. Finally, most children of Black/African descent will develop hypopigmentation in response to seborrheic and/or atopic dermatitis (Fig. 5.1). Therefore we advocate a ten-step approach to evaluation of hypopigmentation in children summarized in Table 5.2.
Table 5.2
Ten-step approach to the diagnosis of hypopigmentation in childhood
Step 1: History |
Age of onset |
Presence of pigmentary loss in the hair or eyes |
Review of past medical history including structural anomalies, intellectual impairments or learning disabilities, prior dermatitis, systemic illness (e.g., lupus erythematosus), drug exposures (e.g., hydroxychloroquine, antibiotics) |
Personal/family history of seizures, autoimmunity, melanoma, atopy, and hypopigmentation |
Step 2: Examination with and without Wood’s lamp |
Examine the patient in good lighting without a spotlight or halogen light for the skin, mucosa, hair, and eyes |
Review of the skin for other skin findings consistent with diagnoses such as angiofibromas of the face, Koenen’s tumors, or fibrous forehead plaque in tuberous sclerosis |
Highlighting with Wood’s lamp is seen in vitiligo, but may be noted in other conditionsa |
Step 3: Identification of lesion morphology |
Confetti-like |
Ovoid or annular |
Over the joints or in the flexures |
Placement along Langer’s lines |
Placement along the lines of Blaschko |
Step 4: Identification of lesional distribution |
Site |
Localized vs. generalized |
Unilateral vs. bilateral |
Flexural vs. extensor locations |
Linear/Blaschkoid |
Presence of Koebner phenomenon |
Number of lesions |
Step 5: Identification of lesion thickness |
Check for sclerotic changes or atrophy |
Identify subtle hyperkeratosis suggesting plaques |
Step 6: Usage of dermoscopy where needed to identify |
Dermal alterations |
Dilated blood vessels |
Subtle thinning |
Underlying pattern of pigmentation or vasculature (e.g., amelanotic melanoma) |
Hyperkeratosis |
Poliosis in vitiligo |
Step 7: How to identify suspicious lesions that require biopsy |
Buttock, periorificial, atrophic or for suspicion of malignancy or poor response to therapeutic trials (see Step 9) |
Step 8: Further ancillary testing |
Infectious testing: KOH, fungal culture |
Karyotyping/genetic evaluation/genetic testing |
Biopsy for fibroblast testing |
Other referrals: e.g., neurology, ophthalmology |
Step 9: Using treatment trials to identify subtle diseases |
Possible treatment trial of topical mid-potency corticosteroids or tacrolimus |
Step 10: Using suspected diagnosis to determine which blood tests to draw |
Diagnoses
Review of every cause of hypopigmentation is not possible in this chapter. Focus on the most common causes of hypopigmentation is given with specific review by diagnosis (Table 5.1).
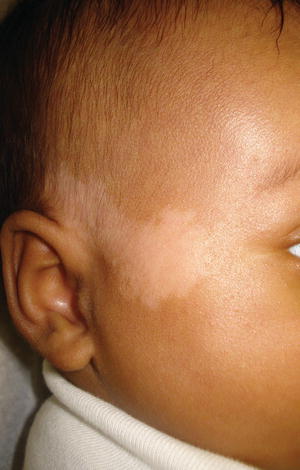
Fig. 5.2
Nevus depigmentosus on the face. This linear lesion was noted at birth. (From the Silverberg NB. Atlas of Pediatric Cutaneous Biodiversity. Spriner 2012, with permission)
Nevus Depigmentosus
Epidemiology
Post-zygotic alterations in pigment genes occur and can generate localized ovoid areas of hypopigmentation termed nevus depigmentosus. Single areas of pigment loss, similar in shape to a café au lait spot, are not uncommon in children of color, especially Hispanic children; however, multiple areas of pigmentary alteration are uncommon and require genetic work-up for genetic and/or chromosomal abnormalities and conditions such as tuberous sclerosis (ash leaf macules, confetti, and thumbprint-like hypopigmentation) [1, 9, 10]. 34.2 % of lesions are seen at birth and most lesions are noted by 3 years of age [1].
Clinical Presentation
Nevus depigmentosus is a localized hypopigmentation usually on the trunk or limbs that does not enhance on Wood’s lamp examination.
Lesions appear anytime from birth to age 2 years and may not become prominent until pigmentary development obviates the lesions.
Nevus depigmentosum is a localized ovoid area of hypopigmentation that is non-enhancing with Wood’s lamp (Fig. 5.2). Lesional appearance may be noted at birth or as late as age 3 years due to delayed pigmentary development. Hypopigmentation of the overlying hairs, the adjacent iris, and the mucosa can accompany a nevus depigmentosus [11]. Lesions are most commonly noted on the trunk, followed by the extremities. Wood’s lamp examination can be used to identify early or subclinical lesions. The mexameter can be used to differentiate the nevus depigmentosus from vitiligo, by relative melanin index [12]. Similarly, in vivo confocal reflectance microscopy can be used to differentiate these lesions from vitiligo [13]. Differentiation from the ash leaf macules of tuberous sclerosis can be difficult and requires a high level of vigilance in screening for the stigmata of tuberous sclerosis. Biopsy shows normal number of melanocytes and reduced pigmentation of the epidermis with clumped melanosomes [5, 14].
Treatment
No treatment exists to remove these lesions other than surgery.
Excimer light sources can temporarily tan lesions to make them less obvious.
>Usage of grafts of pigmented skin or melanocytes can aid in lesional clearance.
No current therapy for permanent removal, other than surgery, exists. Suction blister grafts and melanocyte keratinocyte grafts have been described to help clear lesions [15, 16]. Excimer laser has been described to reduce the prominence of these lesions [17]. A treatment trial with excimer laser can produce alterations on mexameter that differentiate such lesions from vitiligo. Pigmentary differences may become less marked with time in some children and disappearance can occur [18].
Menkes Kinky Hair Carrier
Epidemiology
Menkes kinky hair disease is a rare X-linked recessive defect in copper transport gene ATP7A associated with generalized hypopigmentation and neurologic degeneration [19]
Clinical Presentation
Female carriers, due to random X inactivation, can present with localized or flag-like areas of hypopigmentation and variable neurologic defects [20].
Treatment
No treatment exists for the therapy of the carrier state of Menkes disease.
Prenatal diagnosis can be performed to prevent transmission of the gene when the mutation has been identified in the ATPA7 gene [21].
Lichen Striatus
Epidemiology
Lichen striatus is a common, self-limited condition of childhood
Lichen striatus is a common, self-limited condition of childhood seen in children of all ages and ethnicities and occurring year round. In a survey of 113 Indian children seen for hypopigmentation, 2 children had lichen striatus [1].
Clinical Presentation
Lichen striatus is typified by two phases of disease, a papular/eczematous phase [22] with a spreading, erythematous to flesh-colored plaque occurring along the lines of Blaschko
A second phase characterized by prolonged postinflammatory hypopigmentation in children of color is noted upon clearance of the first stage
Lesions are most common on the lower extremities
Lichen striatus can be seen in children of all ages after infancy and follows a classical clinical course beginning with raised papular lesions that are erythematous to flesh colored along the lines of Blaschko, starting usually distally and progressing proximally along the lower extremity. It is usual for only one segment to be involved, but multiple segment involvement can occur [23–25]. Involvement of the nail with a non-scarring, self-limited dystrophy can be noted [26]. No seasonality is noted in larger studies. Upon resolution of the first phase, which usually takes 6 months but can rarely persist for 3–4 years, hypopigmentation will be noted in most children of Fitzpatrick types IV or higher (Fig. 5.4), with erythema as a sequela in some Asian children. Facial lichen striatus may resolve more rapidly according to one inner city study from the United States [27]. One case has been reported after allogeneic bone marrow transplant [28].
Treatment
Natural clearance is expected without scarring; therefore, therapy is not required.
Therapies can be used to reduce pruritus or erythema or for cosmesis.
Pigmentary Mosaicism/Hypomelanosis of Ito
Epidemiology
Linear pigmentation along these embryological lines is termed pigmentary mosaicism
When associated with structural or developmental anomalies, pigmentary mosaicism is termed hypomelanosis of Ito.
The incidence of pigmentary mosaicism is unknown, but it is not rare, while hypomelanosis of Ito is uncommon, occurring in only 20–30 % of individuals with pigmentary mosaicism. There is no race or ethnicity more likely to be affected; however, due to lyonization, females may be more likely to manifest pigmentary mosaicism (Fig. 5.3).
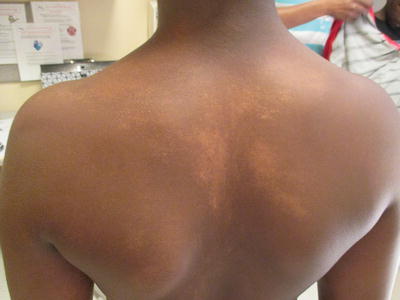
Fig. 5.3
Linear hypopigmentation after flattening of the papular phase of lichen striatus
Clinical Presentation
Hypopigmentation along the lines of Blaschko that is Wood’s lamp negative or equivocal
Multiple lesions, in association with structural and/or developmental anomalies, are termed hypomelanosis of Ito
Pigmentary mosaicism usually appears at birth in darkly pigmented children, such as Indian or African-American children, or sometime in the first 2 years of life in Hispanic or Asian children. Mosaicism can be accompanied by hypertrichosis or hypotrichosis (Fig. 5.4).
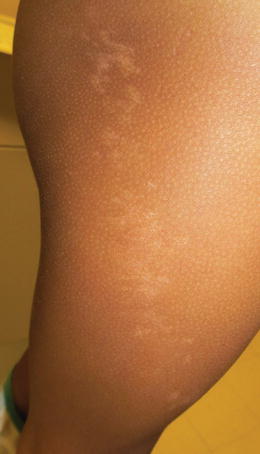
Fig. 5.4
Linear Hypopigmentation in a patten consistent with pigmentary mosaicism
Treatment
No current therapies are available for this disorder; however, judicious clinical monitoring for structural anomalies is recommended
While pigmentary mosaicism cannot be reversed, clinical monitoring is required developmentally in growth, limb lengths, and/or physical or speech development. Cardiac development, brain structure, and ocular development should be monitored.
Incontinentia Pigmenti
Epidemiology
X-linked dominant disorder that is rare
Incontinentia pigmenti (IP) is an X-linked dominant disorder that only rarely affects living male children. It results from mutations in the nuclear factor-ΚB (NEMO/IKBKG) [32].
Clinical Presentation
Four stages of development: vesicular, verrucous, hyperpigmentation, and hypopigmentation
Dental, neurologic, and ocular anomalies can accompany the cutaneous features
IP is apparent at birth or within a few weeks of birth. There are four well-recognized clinical stages of IP. Stage I is apparent at birth or up to 2 weeks of age and consists of linearly arranged vesicles, bullae, erythematous papules, pustules, and erythematous macules. Stage II of IP occurs between week 2 and week 6 of life consisting of linearly arranged verrucous papules. Hyperpigmentation along the lines of Blaschko characterize Stage III beginning at 3–6 months of age. Stage IV consisting of whorled hypopigmentation +/− atrophy, usually, is apparent by the second to third decade of life [32, 33].
Treatment
No current therapy exists for IP
No current treatment exists for IP and treatment is not required clinically. Children who survive infancy have a good prognosis.
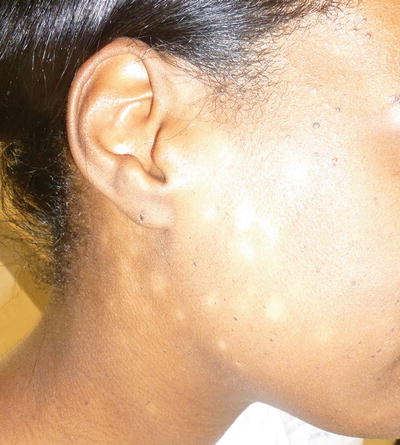
Fig. 5.5
Progressive macular hypomelanosis
Nutritional Deficiencies Causing Hypopigmentation (Table 5.1)
Kwashiorkor
Protein-energy malnutrition (PEM) is the most common cause of nutritional deficiency worldwide. The two main forms are marasmus and kwashiorkor.
Kwashiokor is defined as total body weight of 60–80% that expected for age and height with either edema and/or hypoalbuminemia.
Kwashiorkor is due to insufficient protein intake, despite sufficient caloric intake, and is often seen in children with diets consisting entirely of rice or rice-based beverages. The children often develop failure to thrive, edema, and moon facies, as well as, marked muscle atrophy, hepatomegaly, and a distended abdomen with dilated intestinal loops. Frequent superimposed bacterial and fungal infections often complicate this condition. While PEM is rarely seen in developed countries, Lui et al. reported 12 cases of kwashiorkor diagnosed in the US in children without chronic medical illness. Six of the twelve cases were the result of poor nutritional education, fad diets, presumed food allergy and/or specific food avoidance.
Cutaneous manifestations include generalized hypopigmentation, dyschromia, pallor and xerosis. In mild cases, a superficial desquamation resembling flaking paint (“enamel paint spots”) can be seen, while in more severe cases, confluent areas of eroded skin can occur. The hair in these patients can become thin, dry and abnormally pigmented, with a reddish tinge or occasionally even grey/white. The hair can also develop bands of light and dark colors, referred to as a “flag sign” which represents alternating periods of malnutrition and improvement of nutritional status. Clinical diagnosis should be confirmed with more objective markers including: easily detached hair, edema, skin fissures, faulty wound healing and skin anergy, as well as, albumin < 2.8 g/dl, transferrin < 150 mg/dl or total iron binding capacity < 200mcg/dl. Leukopenia can also be seen due to decreased cellular immunity.
Fad diets, restrictive diets and eating disorders can also result in malnutrition and ultimately kwashiorkor. As in classic cases of kwashiorkor as described above, protein-energy malnutrition from diets can lead to similar cutaneous findings, such as xerosis and pigmentary changes of the skin and hair, and may even be the first appreciable signs of an eating disorder. A 2009 report described a 28 year old woman who presented for evaluation of scaly skin rash and lightening of hair color from brown to blond over the course of 4 years. After an extensive GI evaluation was performed, the patient was ultimately found to have combined vitamin and mineral deficiencies as well as kwashiorkor in the setting of anorexia nervosa and alcohol abuse.
Chronic malabsorptive states, such as those secondary to inflammatory bowel disease, celiac disease, pancreatic insufficiency and post GI surgery, can all lead to nutritional deficiencies. Protein-energy malnutrition has been reported in each of these settings resulting in kwashiorkor with associated findings of xerosis and hypopigmentation. Kwashiokor has been reported in the setting of infantile Crohn’s disease with reversal of symptoms noted 2 weeks after treatment with prednisolone, azathioprine and nutritional supplementation. A 2011 case report of a Type 4/5 dark skinned patient with Kwashiokor secondary to a Whipple procedure used to treat chronic pancreatitis with pseudotumor at the head of the pancreas, demonstrated marked hypopigmentation of the skin and hair as well as ichthyosiform, xerotic skin. The patient was treated with a combination of nutritional support (hypercaloric oral diet, hyperproteic and with medium-chain triglycerides), vitamin supplements, oligoelements and oral replacement of pancreatic enzymes and within 3 months his pigmentation had returned to essentially normal.
Treatment
Treatment must be undertaken with caution, with a focus on correcting electrolyte abnormalities and treating infection. Malnutrition must be corrected with both balanced nutrition and increased caloric intake. Emollients can be used on the skin and cutaneous lesions tend to improve with treatment of the malnutrition.
Vitamin/Mineral/Enzyme Deficiencies
Vitmain deficiencies have also been implicated in hypopigmentation. A case of hyperpigmented macules on the body, longitundal hyperpigmented streaking on the nails and gingiva hyperpigmentation along with diffuse whitening of the hair in a 55 year old female was seen in the setting of Vitamin B12 deficiency. These pigmentary alterations were reversed in 2-3 months, with correction of her Vitamin B12 deficiency. Additionally, Extensive confetti like vitiligo has been described in association with extremely deficient levels of vitamin D.
Selenium deficiency, though rare, can be seen in cases of total parenteral nutrition (TPN), chronic bowel disease and following gastric bypass surgery. It can also be seen in people who survive on diets derived entirely from selenium deficient soil.
Postinflammatory Pigmentary Alteration
Epidemiology
Postinflammatory pigmentary alteration is a leading chief complaint for patients of color.
While the incidence is unknown in children, virtually all children of color will experience postinflammatory pigmentary alteration.
Postinflammatory pigmentary alteration is a pigmentary alteration of the skin that is seen in children of color from early infancy through adolescence. Causes of dyspigmentation include primarily hypopigmentation early on, with the leading insult being seborrheic dermatitis and atopic dermatitis (Fig. 5.1). With increasing age, hyperpigmentation is a common postinflammatory change, seen after bug bites, acne, and eczema lesional clearance.
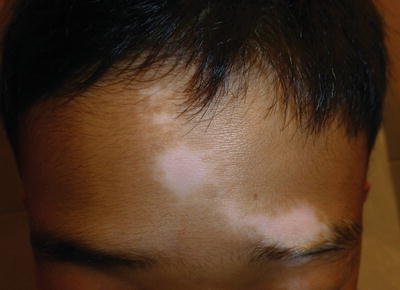
Fig. 5.6
Segmental vitiligo of the forehead
Clinical Presentation
Discrete, well-demarcated areas of pigmentary alteration, either hypopigmentation or hyperpigmentation, are noted.
Pigmentary alterations should be self-limited, but cantake years to resolve in darkly pigmented individuals.
Treatment
Resolution without therapy is expected in most cases
Therapies include treatment of the underlying cause and sun protection
Bleaching, chemical peels, and fraxel laser can be used in older patients to speed resolution.
Waardenburg’s Syndrome
Epidemiology
Waardenburg’s Syndrome is inherited via autosomal dominant inheritance and is characterized by four subtypes.
Waardenburg’s Syndrome types I and III are due to mutations in the PAX3 transcription factor gene. The PAX transcription factor is a key component in neural crest differentiation, melanoblast activation, melanoblast migration, and structures involving the facial bone, facial cartilage, and inner ear. Waardenburg’s Syndrome type II is due to a mutated melanocyte transcription factor, MITF gene. Type IV is due to mutations in SOX10 transcription factor, endothelin-3 gene, and endothelin receptor which all affect neural crest development [32].
Clinical Presentation
The main skin finding in Waardenburg’s Syndrome is depigmented patches with overlying hyperpigmented patches, i.e., islands of repigmentation. Other clinical features may include a white forelock, synophrys, dental caries, broad nasal root, dystopia canthorum (not in type II), heterochromia iridum, congenital sensorineural hearing loss (most common in type II), and Hirschsprung’s disease (type IV) [32, 34, 35]. Cleft lip and palate may be seen in type I and musculoskeletal defects of the upper limb and pectoral muscles may be seen in type III [32].
Treatment
There is no cure for Waardenburg’s Syndrome.
Patients should be evaluated by specialists who can address the known potential characteristics of the syndrome.
Pregnant mothers who have a family history of Waardenberg’s syndrome should take folic acid supplementation due to reported familial cases of neural tube defects.
Waardenburg’s syndrome patients should be screened by specialists for the known features of the syndrome. Hearing screen, rectal examination and genetic testing/counseling are reasonable screening tests for Waardenburg’s syndrome patients. There have been reports of neural tube defects in Waardenberg’s syndrome patients. Folic acid supplementation, though recommended for all women of child bearing age, may be of particular importance in this at risk group [36].
Piebaldism
Epidemiology
Piebaldism is a rare autosomal dominant disorder caused by mutation in the KIT proto-oncogene.
Piebaldism is caused by abnormal melanocyte migration and melanosome transfer, resulting in white forelock and depigmentation over the abdomen and circumferentially on the extremities.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


