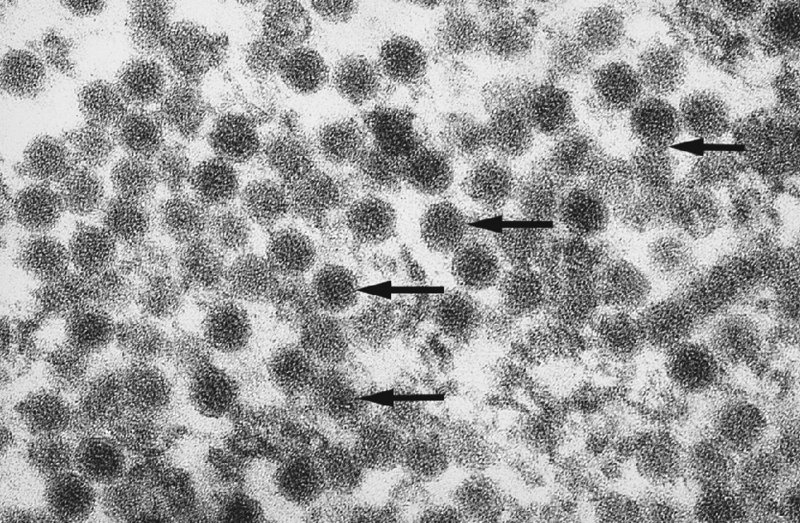
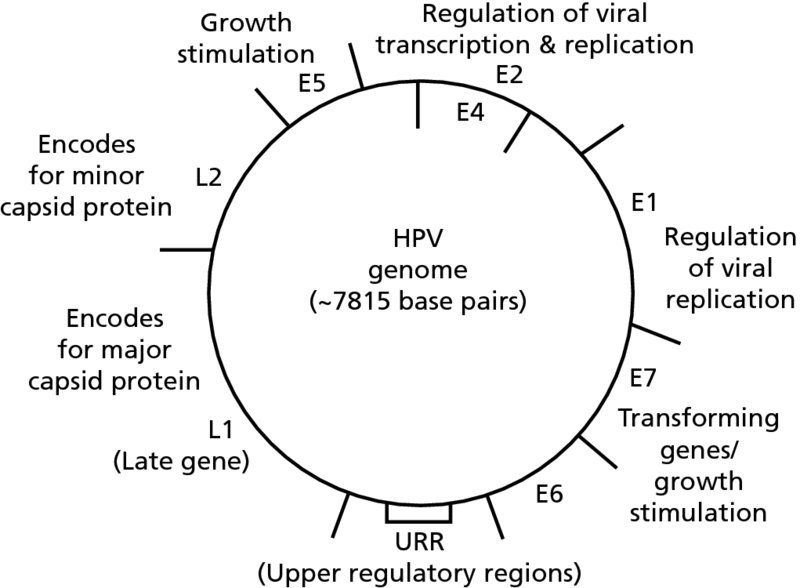
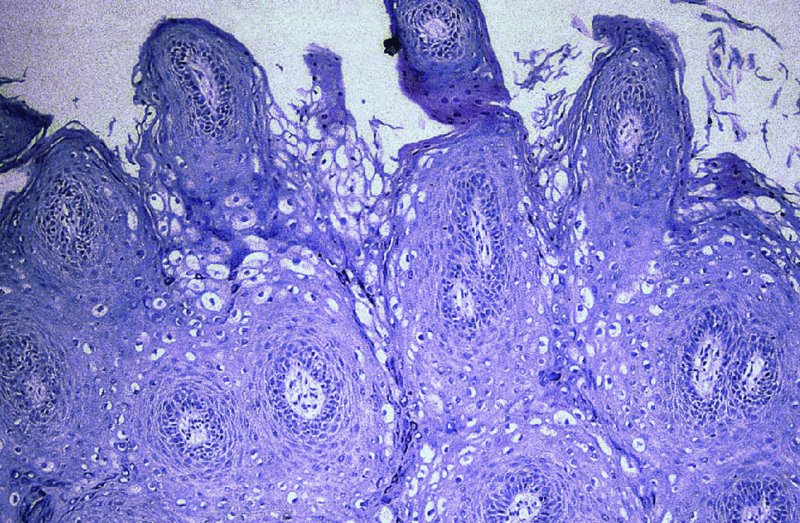
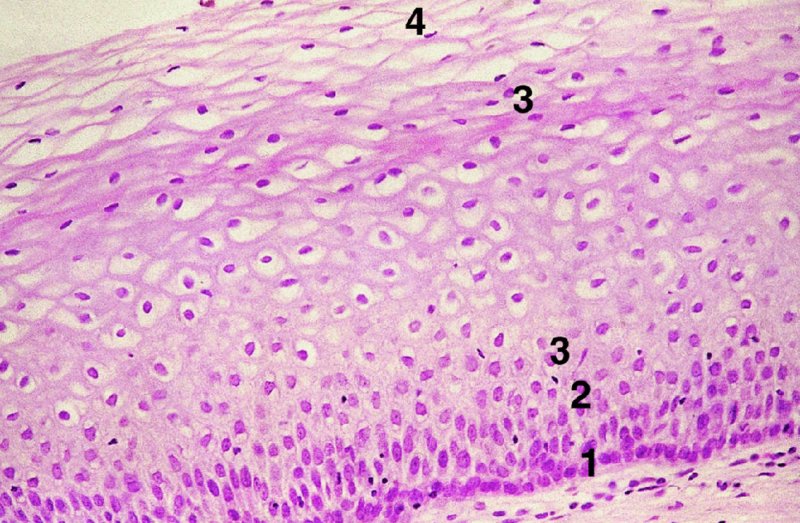
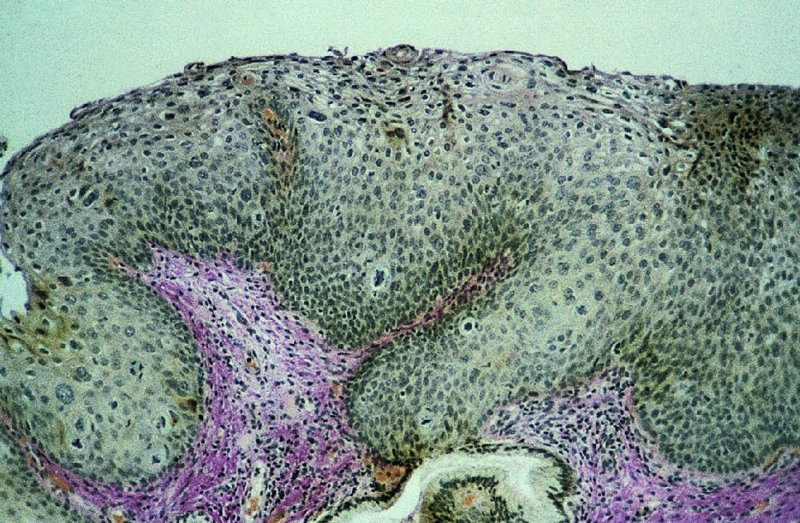
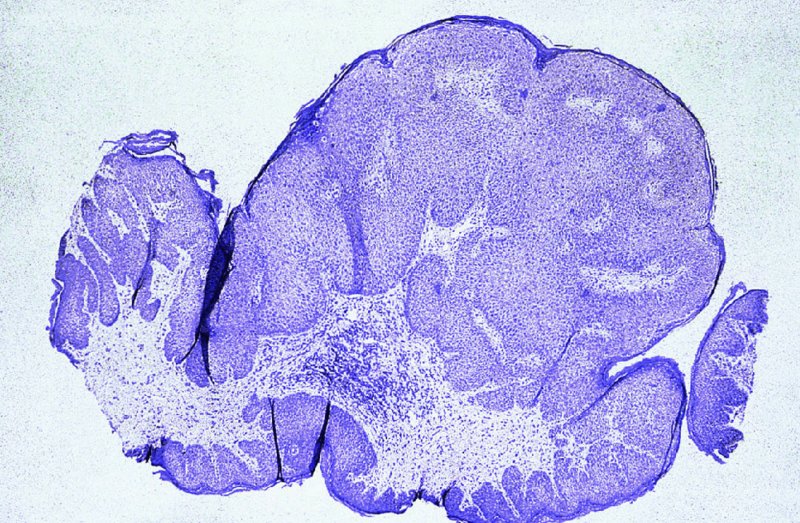
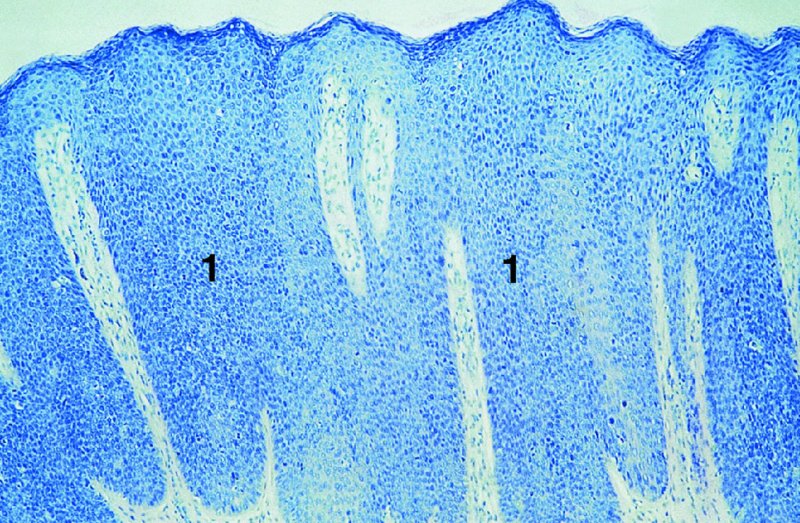
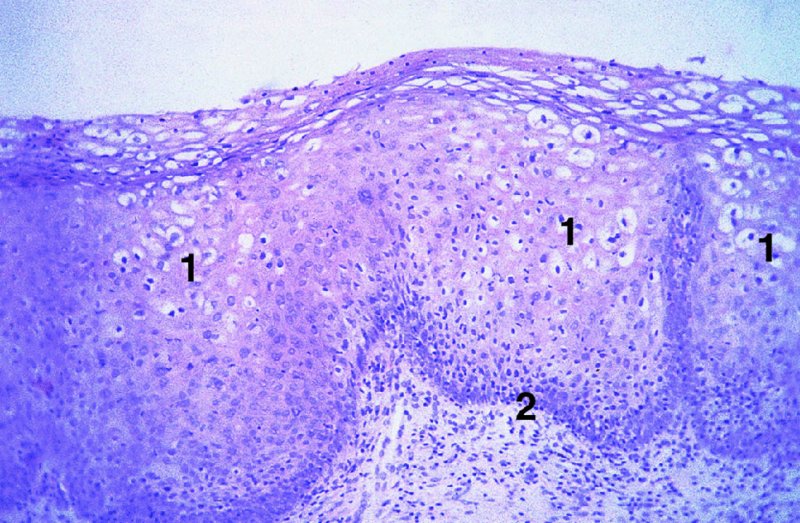
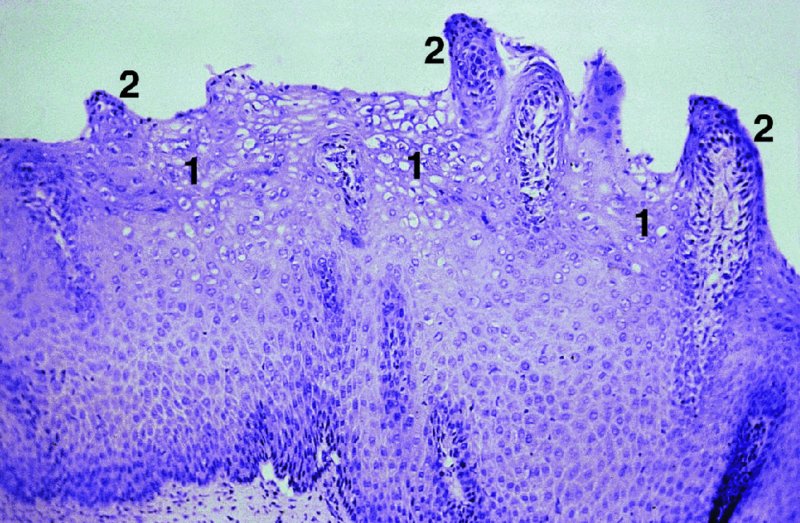
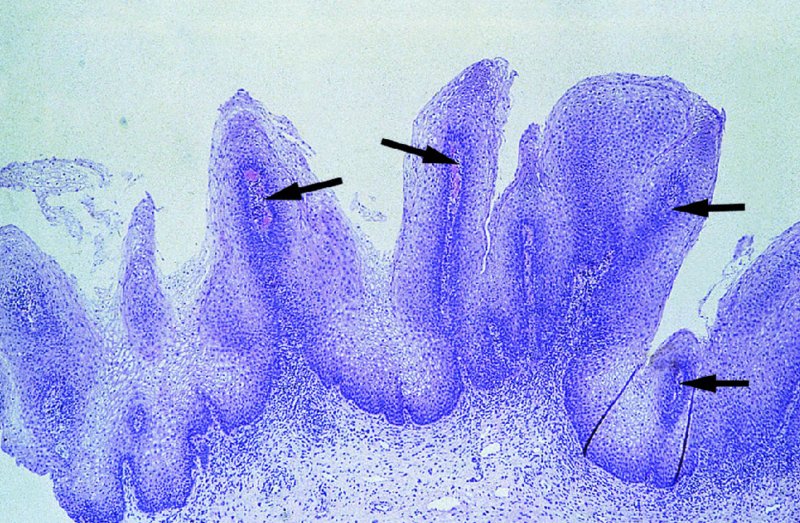
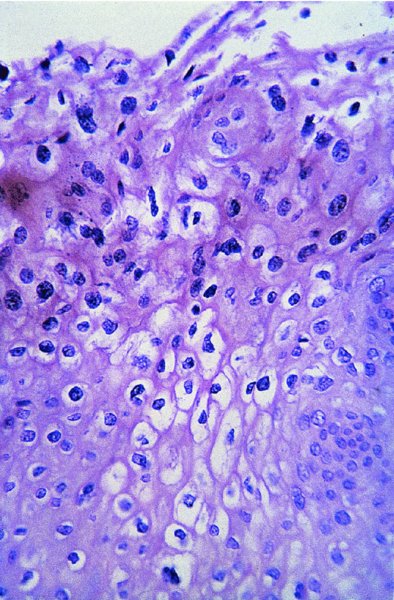
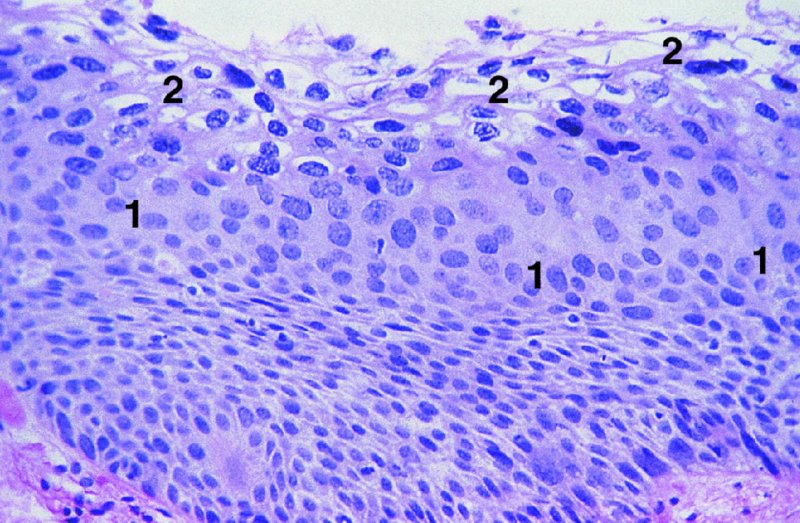
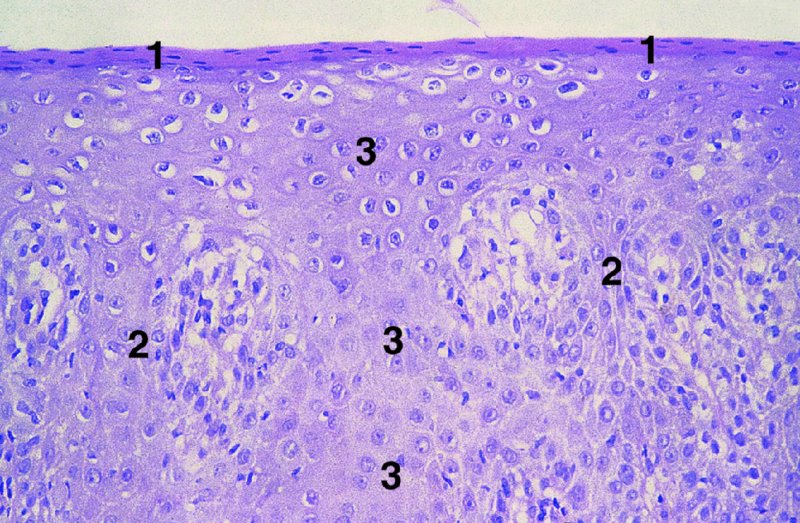
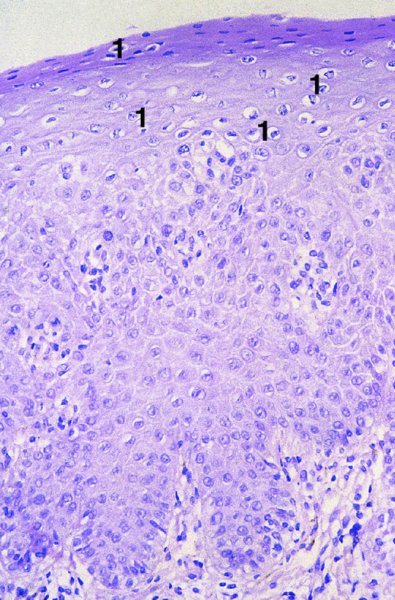
CHAPTER 2 Since 1978, it has been well recognized that human papillomavirus (HPV) infections are frequently associated with genital precancerous and cancerous lesions, namely cervical intraepithelial neoplasia (CIN), vulval intraepithelial neoplasia (VIN), vaginal intraepithelial neoplasia (VAIN), penile intraepithelial neoplasia (PIN), anal intraepithelial neoplasia (AIN), and invasive squamous cell carcinomas. Because viruses are obligate intracellular parasites, they cannot survive extended periods outside their target cells. However, they must be sufficiently stable in the environment to keep alive until they find a susceptible host. The first step in viral pathogenesis is to enter that susceptible host by using any of the available portals of entry, including the skin, mucosa of the eyes, oral cavity, upper gastrointestinal tract, respiratory tract, urogenital mucosa, or anal canal. HPV is a DNA virus that has a double-stranded, tightly coiled, circular genome of about 8000 Da. When the capsid is fully assembled, it forms an icosahedral structure. A complete virion measures approximately 55 nm in diameter (Figure 2.1). HPV has been classified into more than 100 types by DNA homology. The genetic structure of individual types is similar and the genome can be subdivided into three regions: Figure 2.1 Transmission electron microscopic view of papillomavirus particles (arrowed). (Original magnification ×100 000.) Figure 2.2 Schematic drawing of the human papillomavirus genome organization. It is generally assumed that the initial HPV infection in the squamous epithelium requires that the virus be in the form of a complete virion, i.e., a central core of DNA surrounded by its protein coat. Once it enters the host, for example through the genital mucosa, the steps of viral pathogenesis within the individual target cells include the following: adsorption, penetration, uncoating, transcription, translation, replication, assembly, and release. The replication of HPV is closely geared to the differentiation of the cervical epithelium. The initial infection occurs in the basal layer of undifferentiated stem cells. At this stage, only early HPV proteins are synthesized. These include: The synthesis of the early proteins is switched off as the cells are displaced upwards through the epithelium and start to differentiate. The viral structural proteins, L1 and L2, are synthesized in the superficial epithelium prior to exfoliation and are assembled into capsids enclosing viral DNA. The progeny virions are released from the infected cells and this is accompanied by cytoskeletal collapse induced by another viral gene product, E4. Infected cells may not be able to differentiate fully as a result of either (i) functional interference of cell cycle-regulating proteins, caused by viral gene expression, e.g., interaction between HPV16 E6 with cellular protein p53, interaction between HPV16 E7 with cellular retinoblastoma tumor suppressor protein (pRB), or (ii) overproduction of E6 and E7. When these events occur the host DNA synthesis continues unchecked and leads to rapidly dividing undifferentiated cells with morphologic characteristics of high-grade epithelial disease. It is thought that accumulated chromosomal breakages, rearrangements, deletions, and other genomic mutations in these cells lead to cells with invasive capability and ultimately to cervical malignancy. It was agreed in 1979 that HPVs would be named by number, and in their order of discovery. On the basis of type-specific odds ratios and prevalence of types by case or control status, 15 HPV types have been deemed as oncogenic or high risk (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82), three types are considered as probable high-risk types (26, 53, and 66), and 12 types are classified as low risk (6, 11, 40, 42, 43, 44, 53, 54, 61, 70, 72, 81). The World Health Organization (WHO) has declared HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 66 as class I carcinogens for humans. Successful transmission of genital HPV infections seems to depend upon the access of the virus into epithelial cells capable of dividing. The only such cells in the squamous epithelium are those of the basal layer. Thus, HPV infection occurs upon exposure of the basal cells to infectious virus particles as a result of minor traumas of the epithelium, e.g., as a result of sexual intercourse. After an established viral infection, two possible pathways may ensue: In the latter case, HPV stimulates the proliferation of the basal cells (and the synthesis of early HPV proteins as discussed above), leading to the formation of a visible epithelial lesion, e.g., a genital wart. In such an active replicative infection, the number of viral particles increases substantially during the life cycle of the epithelial cell until the cell containing large numbers of complete virions is exfoliated at the epithelial surface. As with many other viruses, this productive HPV infection is associated with viral cytopathic effects (CPE), and it is these CPEs that create the typical histologic and cytologic changes recognizable as morphologic manifestations of an HPV infection on light microscopy. HPVs do not significantly deviate from most other viruses in that they are clearly capable of inducing clinical, subclinical, and latent infections. Unfortunately, there is some confusion in the literature concerning the definitions of these three manifestations. In particular, the term subclinical HPV infection has become obsolete. In the past this used to mean different things by different authors and created a lot of confusion. Clinical HPV infection causes low- or high-grade precancers (CINs or squamous intraepithelial lesions), in which the lesion should be readily apparent by all the generally used clinical diagnostic means, i.e., colposcopy. Low-risk HPVs, especially types 6 and 11, are associated with mainly exophytic genital warts (Figure 2.3), and cellular features are quite distinct when compared with a normal squamous epithelium (Figure 2.4). Flat or endophytic lesions are mainly caused by high-risk HPVs and may be associated with CIN (Figures 2.5–2.12). Although low-risk HPV infections cause subtle changes at the cytoplasmic and nuclear level (Figures 2.13, 2.14), it is not possible to distinguish them reliably from high-risk HPVs by histologic appearances. Figure 2.3 A typical benign exophytic condyloma acuminatum. The lesion is characterized by extensive papillomatosis and koilocytosis, as well as a thin layer of dyskeratotic (parakeratotic) superficial cells. No signs of dysplasia are present. Figure 2.4 Normal squamous epithelium of the ectocervix. All distinct cell layers (basal (1), suprabasal (2), intermediate (3), and superficial (4)) are clearly distinguished in this well-glycogenated epithelium. Figure 2.5 A typical endophytic condyloma associated with cervical intraepithelial neoplasia 2 (human papillomavirus CIN2). The acanthotic epithelium shows a dysplastic change extending to one-half to two-thirds of the epithelial thickness, with some highly abnormal (multipolar) mitotic figures. The uppermost one-third of the epithelium is occupied by characteristic koilocytes. This abnormal epithelium shows an endophytic growth pattern, extending down to underlying connective tissue and endocervical glands, giving the lesion an inverted appearance. Figure 2.7 A classical Bowenoid papulosis lesion of the vulva. This acanthotic focus of epithelium is flat, giving the gross appearance of a macule. The full thickness of the epithelium is composed of highly abnormal cells of the basaloid type (1), and extensive mitotic activity is present. No cytopathic effects of human papillomavirus (HPV) are detectable, but, using in situ hybridization, this lesion contained HPV16 DNA, localized in scattered cells of the most superficial layers. (Toluidine blue.) Figure 2.10 Yet another distinct morphologic manifestation of human papillomavirus infection, known as spiked condyloma, in the vagina. This lesion, extending laterally on the vaginal wall, has a spiked appearance on colposcopy (see Chapter 12) because of the numerous papillary structures covered by koilocytotic squamous epithelium and supported by a connective tissue core with abundant blood vessels (arrowed). Notice a completely different growth pattern than that of the lesion depicted in Figure 2.7. Figure 2.11 A medium-power detail from the superficial layers of a highly proliferative flat condyloma to illustrate the typical appearance of the koilocytotic cells. Marked nuclear changes are obvious, characterized by increased size, variable shape, hyperchromasia, binucleation, and karyopyknosis (in most superficial cells). These abnormal nuclei are surrounded by extensive cytoplasmic clearing (koilocytosis), giving the typical light microscopic morphology. Figure 2.12 Another flat human papillomavirus lesion associated with cervical intraepithelial neoplasia 2 (HPV CIN2). One-half to two-thirds of the epithelial thickness is occupied by a basaloid type of transformed cells (1), with few mitotic figures. There is a sharp border between this dysplastic layer and the uppermost one-third of the epithelium, which is composed of extensive koilocytosis (2). This is a frequent finding in these flat HPV CIN lesions. Figure 2.13 This and the next photomicrograph (Figure 2.14) demonstrate a human papillomavirus (HPV) infection. In this biopsy from an acetowhite lesion on the introitus, a thin layer of superficial (1) parakeratotic cells is seen, responsible for the acetowhite reaction. The epithelium is acanthotic, extending as narrow streaks (2) downwards. The intermediate layer is composed of squamous cells (3) with varying degrees of cytoplasmic vacuolization. In contrast to true koilocytes, however, these vacuolized cells lack the nuclear abnormalities shown in Figure 2.11. The nuclear size is not altered compared with that of the non-vacuolized counterparts, and the nuclei have retained their normal staining properties (i.e., they are not hyperchromatic). This lesion was caused by HPV type 11. Figure 2.14 Another biopsy from the vagina, showing subtle epithelial (1) changes consistent with a subclinical human papillomavirus (HPV) infection. Observable changes are very much like those in Figure 2.13. Again, cells with vacuolized cytoplasm are seen, but they lack the nuclear abnormalities of true koilocytes. This lesion was caused by HPV type 42. Latent viral infections represent cases where viral genome (DNA or RNA) is present in an otherwise normal target tissue. Clinical, subclinical, and latent HPV infections are the most common sexually transmitted viral diseases today. Asymptomatic HPV infection can be detected in 5–20% of sexually active women of reproductive age. The assessment of whether epithelium is normal or not should always be based on careful histopathologic and molecular examination. This is often as a result of a directed punch biopsy. There is little doubt that HPV genomes may persist in epithelial cells without inducing any cytopathic changes. HPV DNA has been identified in cervical smears from colposcopically and cytologically healthy women. Latent infections which are presumed to be non-infectious probably represent the largest reservoir for genitoanal HPV infections. It is speculated that periodic activation and phenotypic expression of small infectious foci within the field of latent infection could represent a source of HPV for most of the sexually transmitted HPV-associated diseases. So far it is not clear whether these latent infections may also be residuals of the lesions having regressed spontaneously. It has also been suggested that reactivation of a latent infection in association with possible immune senescence may explain the rise in HPV prevalence in sexually active women in the fifth and sixth decades of life. The prevalence of HPV in women with normal cytology at any given point in time is approximately 10%. HPV16 is consistently the most common type worldwide, contributing to 50–55% of invasive cervical cancer cases. A further contribution of 15–20% comes from HPV18 and HPV45. Bosch et al. reported that the pattern of HPV DNA prevalence by age groups is similar to the patterns of HPV incidence. Rates of exposure in young women are high and often include multiple types. There is a spontaneous and rapid decrease of the HPV DNA detection rates in the middle-age groups followed by a second rise in the postmenopausal years. In women with normal smears the prevalence of all HPV types is age related, decreasing from 20% in women aged between 20 and 25 years to 4.5% in women aged over 30 years. High-risk HPV types decrease from 7% in women aged between 20 and 25 years to less than 2% in women aged over 30 years. In those women aged over 30 years with normal cytology, the HPV clearance and acquisition rates have been estimated at 30–40% and 0.5% per year, respectively. HPV is virtually absent in women with no previous sexual experience, i.e., in virgins. With increasing sexual experience, the risk of contracting HPV infections seems to increase. The number of sexual partners is one of the most significant risk factors for genital HPV infections. This risk increases in parallel with the number of sexual partners. To interfere with the sexual transmission of HPV, it is of prime importance to assess the reservoirs of this virus in both sexes. Epithelium of the male genital tract (i.e., penis, urethra, and prostate) acts as a reservoir, as HPV DNA exists in apparently normal mucosal sites of the penis, urethra, and prostate. The existence of such a reservoir, in turn, is the prerequisite for sexual transmission of HPV, and for the subsequent detection of HPV-associated precancer lesions and identical HPV types in both sexual partners. Elucidation of the risk factors for the etiologic agent (HPV) is mandatory to identify the populations at risk for cancer precursors. Epidemiological studies indicating that HPV infection with oncogenic viral types is far more common than cervical neoplasia suggest the necessity of cofactors in cervical carcinogenesis. The long time lag between initial infection and eventual malignant conversion suggests that random events may be necessary for such conversion, and the spontaneous regression of many primary lesions suggests that most patients are not exposed to these random events. The major risk factor for genital HPV infections is sexual behavior, particularly multiple sexual partners. Cofactors in the form of cigarette smoking, oral contraceptives, and pregnancy are well documented. Almost irrespective of the target population, the risk factors predisposing women to this infection remain the same, with some minor modifications.
Human papillomaviruses in the pathogenesis of lower genital tract neoplasia
2.1 Introduction
2.2 Characteristics of human papillomaviruses
2.3 Manifestations of genital human papillomavirus infections
Clinical infections (including benign condylomata)
Latent human papillomavirus infections
2.4 Prevalence of genital human papillomavirus infections
2.5 Transmission of genital human papillomavirus infections
Sexual transmission
2.6 Risk factors for genital human papillomavirus infections
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


