Human Immunodeficiency Virus
Howard Minkoff
Human Immunodeficiency Virus
The HIV epidemic is a quarter of a century old and in that time frame has transformed from a uniformly lethal disease of unknown etiology to a manageable chronic disease, albeit one with complicated treatment regimens and mortality remaining an inherent risk. The success of treatment regimens has led to the emergence of two epidemics—one among those with access to highly active therapies as well as a larger one among those without. In this chapter, the pathophysiology and epidemiology of HIV will be summarized, and the management of obstetric and gynecologic patients who are infected will be detailed. Given the ability of U.S. providers to avail themselves of the most efficacious therapies, they should be able to assure their patients that perinatal transmission of HIV will be an extremely uncommon event and that gynecologic morbidities can be minimized.
Microbiology
HIV is a lentivirus, from the family of retroviruses, which characteristically have an RNA genome contained within a capsid and a lipid envelope. Retroviruses constitute a large and diverse family of enveloped RNA viruses that use the transcription of virion RNA into linear double-stranded DNA as a replication strategy, with subsequent integration into the host genome. The characteristic enzyme used for this process, an RNA-dependent DNA polymerase that reverses the flow of genetic information, is known as reverse transcriptase. The unique lifestyle of the retrovirus involves two forms, a DNA provirus and an RNA-containing infectious virion. Infection is initiated by the binding of a protein on the surface of the virus (gp120 Env protein) to the CD4 molecule found on some T-cells, macrophages, and microglial cells. CD4 was first shown to be a viral receptor in a number of studies showing the susceptibility of CD4-bearing cells to infection and the ability to block infection with anti-CD4 monoclonal antibodies in culture.
HIV is composed of core (p18, p24, and p27) and surface (gp120 and gp 41) proteins, genomic RNA, and the reverse transcriptase enzyme surrounded by a lipid bilayer envelope. The virion contains three structural genes (gag, pol, and env) as well as a complex set of regulatory genes (including tat, vif, nef, vpu, and ref) that control the rate of virion production. As noted, it preferentially infects cells with the CD4+ antigen, particularly helper lymphocytes but also macrophages, cells of the central nervous system, and according to some evidence, cells of the placenta. At least two other cell surface molecules help HIV enter cells. These coreceptors for HIV, called CXCR4 and CCR5, are receptors for chemokines. Individuals who are homozygous for a deletion at the CCR5 gene appear less likely to acquire HIV, while deletion heterozygotes progress less rapidly if infected. On the basis of cell tropism, HIV strains can be broadly divided into two categories—macrophage-tropic (M-tropic) and T-cell tropic (T-tropic). M-tropic strains use CCR5 as a coreceptor and are referred as R5 viruses. They primarily infect macrophages and primary T cells and infect
poorly CD4+ T-cell lines. In addition, these viruses tend to be transmitted sexually more easily. T-tropic strains use the CXCR4 coreceptor, which is most expressed in CD4+ T cells. Also referred to as X4 viruses, they induce the formation of syncytia in the infected cells. Early in the course of HIV infection, the R5 strain viruses predominate; however, eventually, both X4 and R5 strains are recovered (Fig. 20.1).
poorly CD4+ T-cell lines. In addition, these viruses tend to be transmitted sexually more easily. T-tropic strains use the CXCR4 coreceptor, which is most expressed in CD4+ T cells. Also referred to as X4 viruses, they induce the formation of syncytia in the infected cells. Early in the course of HIV infection, the R5 strain viruses predominate; however, eventually, both X4 and R5 strains are recovered (Fig. 20.1).
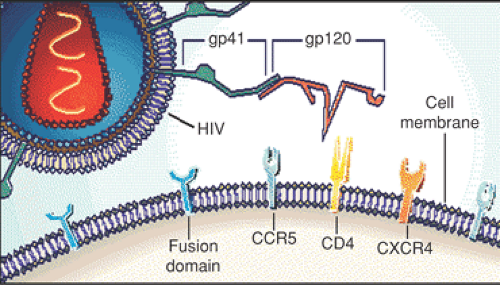 Figure 20.1 Early in the course of HIV infection, the R5 strain viruses predominate, but eventually both X4 and R5 strains are recovered. (From Levy JA. Infection by human immunodeficiency virus—CD4 is not enough. N Engl J Med 1996;335:1525–1527 , Figure 1A. Illustrations © Massachusetts Medical Society, with permission.) (See Color Plate) |
Epidemiology
By the end of 2003, approximately 1,039,000 to 1,185,000 persons in the United States were living with HIV/AIDS, an estimated 24% to 27% of whom were unaware of their infection, with approximately 25% being women. AIDS cases increased rapidly in the 1980s and peaked in 1992 (an estimated 78,000 cases diagnosed that year) before stabilizing in 1998; since then, approximately 40,000 AIDS cases have been diagnosed annually. Over the course of the epidemic, even before effective treatments were widely utilized, the number of AIDS cases decreased 47% from 1992 to 1998. The majority of AIDS cases continue to occur among males; however, the proportion of all AIDS cases for females increased from 15% in 1981 to 1995 to 27% in 2001 to 2004. Not coincidently, the vast majority of cases of pediatric AIDS are secondary to vertical transmission of HIV from mother to fetus. The proportion of all AIDS cases attributable to high-risk heterosexual contact (i.e., sexual contact with a person at high risk for or infected with HIV) from 1981 to 1995 was 10% and increased to 30% from 2001 to 2004. Among males and females, case rates among blacks (males: 131.6 per 100,000; females: 67.0 per 100,000) were 7 and 21 times higher, respectively, than rates for whites (males: 18.7 per 100,000; females: 3.2 per 100,000).
Other important trends characterize the epidemic in women. For example, an increasing proportion of AIDS cases are occurring in women in the South, perhaps reflecting the dramatic increase in other sexually transmitted diseases first seen in that region more than a decade ago. Poverty status also might vary with gender, with women substantially more likely to be covered by Medicaid and less likely to be privately insured. These data demonstrate, yet again, that poverty, drug use, and sexually transmitted diseases continue to fuel the HIV epidemic among women in the United States.
Pathophysiology
HIV infection induces a profound immune dysfunction, with abnormalities in every arm of the immune system. While studies of long-term nonprogressors (HIV-infected patients who are asymptomatic and have normal CD4+ T-cell counts in the absence of treatment) have revealed several immune mechanisms that are significant in controlling HIV infection, the virus has several inherent strategies by which to escape this vigorous immune response and to continue replicating. The most studied of these strategies are antigenic variation, down regulation of the surface expression of major histocompatibility complex (MHC) molecules, and reduction of specific CD8+ T cells. Once the immune system has become debilitated, infected individuals are rendered susceptible to opportunistic infections (e.g., Pneumocystis carinii pneumonia [PCP] and central nervous system toxoplasmosis) and neoplasias (e.g., Kaposi’s sarcoma) that rarely afflict patients with intact immune systems. An HIV-infected patient with one of several specific opportunistic infections, neoplasia, dementia encephalopathy, or wasting syndrome is diagnosed as having AIDS. The diagnosis of AIDS can be made in the absence of laboratory evidence of infection if the patient has no other known cause of immune deficiency and has the definitive diagnosis of one of a number of indicator diseases. In 1993, the Centers for Disease Control and Prevention (CDC) changed the case definition to include all individuals with HIV infection whose CD4 counts drop below 200 cells/mm3 as well as HIV-infected individuals with advanced cervical cancer, pulmonary tuberculosis, and recurrent pneumonia.
At the time of initial infection, an individual may be asymptomatic or may develop an acute mononucleosislike syndrome that can be accompanied by aseptic meningitis. There is then an immediate viremia of substantive proportions (up to ten billion viral particles turned over per day) and an equally impressive immune responsive with similar levels of T-cell turnover. Antibodies can be detected in almost all individuals at 6 to 12 weeks after exposure, but in rare circumstances, this latent period (the so-called “window phase”) can be longer. After seroconversion has occurred, an asymptomatic period of variable length usually follows. The median clinical latency in the absence of effective therapy is estimated at approximately 11 years. Very few infected persons (<5%) develop AIDS within 3 years. Evidence of immune dysfunction may be followed by clinical conditions ranging from fever, weight loss, malaise, lymphadenopathy, and central nervous system dysfunction to
infections such as herpes simplex virus or oral candidiasis. These nonspecific conditions usually are progressive and are a prelude to an opportunistic infection that is diagnostic of AIDS. Pre–HAART (highly active antiretroviral therapy) era studies of infected individuals reported that 5 years after infection was confirmed, up to 35% had progressed to AIDS. A study of subjects with hemophilia demonstrated that the incidence rate of AIDS after seroconversion was 2.67 per 100 person-years and was directly related to age (younger individuals developed AIDS at a slower rate). The level of virus in the plasma can provide an estimate of the probability that an individual will develop AIDS within 5 years. It should be noted that almost all of these statistics antedate the use of new, more powerful antiretroviral agents that have a significant effect on both clinical outcomes and surrogate markers of disease progression.
infections such as herpes simplex virus or oral candidiasis. These nonspecific conditions usually are progressive and are a prelude to an opportunistic infection that is diagnostic of AIDS. Pre–HAART (highly active antiretroviral therapy) era studies of infected individuals reported that 5 years after infection was confirmed, up to 35% had progressed to AIDS. A study of subjects with hemophilia demonstrated that the incidence rate of AIDS after seroconversion was 2.67 per 100 person-years and was directly related to age (younger individuals developed AIDS at a slower rate). The level of virus in the plasma can provide an estimate of the probability that an individual will develop AIDS within 5 years. It should be noted that almost all of these statistics antedate the use of new, more powerful antiretroviral agents that have a significant effect on both clinical outcomes and surrogate markers of disease progression.
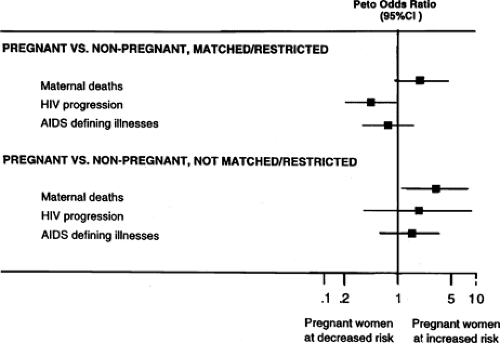 Figure 20.2 Summary odds ratio of studies that attempted to control for confounding by matching or restriction techniques compared with those which had not used matching or restriction. (CI, confidence interval.) (From French R, Brocklehurst P. The effect of pregnancy on survival in women infected with HIV: a systemic review of the literature and meta-analysis. Br J Obstet Gynecol 1998;105:827–835 , with permission.) |
From 1981 to 2004, a total of 522,723 deaths among persons with AIDS were reported to the CDC. In the era of highly active antiretroviral therapy, prognosis has improved even for those with AIDS at initiation of treatment. The time at which 25% of individuals had died after an AIDS diagnosis increased significantly from 0.56 years (95% confidence interval [CI] 0.50 to 0.64) in the no/monotherapy era to 5.08 years (95% CI 2.39 to 10.79) in the HAART era. The proportion of persons living at 2 years after AIDS diagnosis was 44% for those with AIDS diagnosed from 1981 to 1992, 64% for 1993 to 1995, and 85% for 1996 to 2000. Survival for more than 1 year after diagnosis for persons with AIDS diagnosed from 1996 to 2003 was greater among Asians/Pacific Islanders, whites, and Hispanics than among blacks and American Indians/Alaska Natives. Viral load and CD4 counts can be used to predict the likelihood that an individual will develop AIDS during a given follow-up period. At the current time, appropriately controlled studies reveal no convincing evidence that the natural history of HIV infection is influenced by gender or pregnancy (Fig. 20.2). Clearly, medications have improved prognosis dramatically, though it must be recognized that they also have been associated with substantial morbidity (e.g., lipodystrophy).
Management
Monitoring
The guiding principle in the care of HIV-infected pregnant women continues to be strict adherence to the standards of care that apply to all other HIV-infected individuals. The first step in that care is the monitoring of immune status with CD4 counts and viral loads. Checking for viral resistance also has become a key component of monitoring regimens (vide infra). Viral loads can be checked every 3 to 4 months. Once a decision is made to initiate therapy, viral-load monitoring should occur monthly until virus is no longer detectable and then can be cut back to three or four times per year. With appropriate therapy, a drop of 1.5 to 2 logs within 1 month of initiation of treatment can be anticipated. The recommended timing of initiation of therapy (vis-a-vis CD4 counts and viral loads) has undergone several revisions over the last few years. This evolution reflects the awareness that despite the clear benefits of therapy, there also are well-recognized toxicities that make it difficult to maintain strict adherence. Some data suggest that no harm befalls patients who delay therapy until viral loads rise and CD4 counts drop further than had previously been recommended. Current guidelines are to start therapy in nonpregnant women when the viral load is >100,000 copies or when the CD4 count drops below 350 cells/mm3. In the nonpregnant state, when those thresholds are passed, HAART should be initiated. The appropriate guidelines for initiating therapy in pregnancy follow.
Resistance Testing
Resistance testing has become a staple of care for HIV-infected individuals. The viral RNA is reverse transcribed
into complementary DNA (cDNA) by the viral reverse transcriptase through use of a cellular lysine tRNA molecule as a primer; subsequently, the RNAase activity of the reverse transcriptase degrades the viral RNA template. The reverse transcriptase incorporates an incorrect nucleotide every 1,500 to 4,000 bases, which explains the rapid occurrence of mutations. Some of the resulting mutations provide a survival advantage, leading to drug-resistant strains.
into complementary DNA (cDNA) by the viral reverse transcriptase through use of a cellular lysine tRNA molecule as a primer; subsequently, the RNAase activity of the reverse transcriptase degrades the viral RNA template. The reverse transcriptase incorporates an incorrect nucleotide every 1,500 to 4,000 bases, which explains the rapid occurrence of mutations. Some of the resulting mutations provide a survival advantage, leading to drug-resistant strains.
There is accumulating evidence that transmitted resistant mutants may persist for indefinite periods after initial infection, these viral variants may be detectable by standard assays used in clinical practice, the prevalence of resistance in antiretroviral-naive patients is increasing, and baseline resistance may be associated with adverse virologic outcomes. For these reasons, baseline HIV resistance testing is now recommended for all patients with established infection, including pregnant women, prior to initiating treatment.
Most randomized trials of resistance testing have demonstrated that those assigned to study arms with access to resistance test results have a greater reduction in viral load after the initiation of salvage therapy, though follow-up generally has been short. These tests are recommended for individuals prior to commencing therapy or after failed therapy. Treatment failure is defined as the failure to attain an undetectable level of virus or the persistent presence of virus after it has become undetectable. Transient low-level viremia may not be the same as a drug failure, and sustained response to treatment can occur even in the setting of occasional low-level viremia. Blood for testing should be obtained before a failing regimen is discontinued lest wild-type virus overgrow before the test is performed. In that circumstance, an individual with no apparent resistance would still fail therapy when reexposed to drugs that favor the growth of the resistant virus over the wild-type strain. In essence, resistance testing is more useful for ruling out, than for ruling in, therapies to be utilized in a given patient. That is because, as noted, the absence of resistance may merely reflect the reemergence of a wild-type strain after an antiretroviral agent has been withdrawn. In that circumstance, the assays will not detect a low volume of a minority mutant strain. However, if the patient is reexposed to the offending agent, the resistant strain may again attain dominance. It is not possible to perform resistance studies if there are <1,000 copies of detectable virus.
Currently, two types of testing are available—genotypic and phenotypic—each with distinct advantages and disadvantages. Phenotypic testing compares the ability of the virus to replicate in various concentrations of an antiretroviral drug, with its replication in the absence of the drug. In general, if the amount of drug required to inhibit viral production by 50% is fourfold or greater for the patient virus than the control strain, then the patient’s strain is considered resistant. Specific cutoffs for resistance for each drug are based on clinical correlation, and the serum levels usually are attainable for a given drug. Measurement of drug levels per se has not yet been shown to be useful addendums to standard monitoring.
Genotypic testing is directed at detecting mutations in the genes that encode reverse transcriptase and protease formation by the virus. Point mutations in the virus result in the substitution of amino acids in the proteins produced (i.e., reverse transcriptase or protease). The significance of these point mutations has to be determined by correlating specific mutations with phenotypic resistance as measured by viral susceptibility assays and correlation with clinical response to therapy. Genotypic changes leading to resistance are believed to result from the combination of the rapid turnover of HIV (107 to 108 rounds of replication per day) and the high error rate of reverse transcriptase when replicating the nearly 10,000 nucleotides present in the HIV genome. Genetic mutants with resistance to antiretroviral agents are then selected by evolutionary pressure when incompletely suppressive drug regimens are used. The rate at which resistance develops will depend on the number of mutations necessary for a significant change in susceptibility to occur. The genetic basis for resistance must be understood before the impact of a specific mutation can be predicted. In addition, mutational interactions may make prediction of phenotype (i.e., susceptibility) difficult when multiple mutations are present. Prediction of cross resistance to other drugs within a class such as protease inhibitors (PIs) can be difficult to predict based only on genotype. In clinical practice, most clinicians will rely on algorithms developed by panels of experts or to online databases. Obstetricians should interpret and act on these results in consultation with an expert in the field.
During pregnancy, HIV drug-resistance testing is recommended for several subsets of women. All pregnant women who are not currently receiving antiretroviral therapy should be tested before starting treatment or prophylaxis. Additionally, all pregnant women who are receiving antenatal antiretroviral therapy and have virologic failure with persistently detectable HIV RNA levels or who have suboptimal viral suppression after initiation of antiretroviral therapy should undergo resistance testing. While it would be ideal to have results back before starting therapy in pregnancy, it has been recommended that in certain circumstances (e.g., late registrant), empiric initiation of antiretroviral therapy before results of resistance testing are available may be warranted, with adjustment as needed once the results are known. Ideally, the development of resistance would be avoided in the first instance. Several steps can be taken toward that end. The use of highly active antiretroviral combination therapy to maximally suppress viral replication during pregnancy is the most effective strategy to prevent the development of resistance and to minimize the risk of perinatal transmission. Finally, all pregnant women should be counseled about the importance of adherence to prescribed antiretroviral medications to reduce the potential for development of resistance.
Although perinatal transmission of resistant virus has been reported, it appears to be unusual, and there is little evidence that the presence of resistance mutations increases the risk of transmission when current recommendations for antiretroviral management in pregnancy are followed. In studies in which transmitting mothers had mixed viral populations of wild type and virus with low-level zidovudine (ZDV) resistance, only wild-type virus was found in the infants, and other studies have suggested that drug-resistance mutations may diminish the fitness of the virus, possibly leading to a decrease in transmissibility. Neither resistance to NVP that develops as a result of exposure to single-dose NVP nor exposure to single-dose NVP in a prior pregnancy have been shown to affect perinatal transmission rates.
Pharmaceutical Therapy
The standard approach to the medical treatment of HIV infection continues to evolve at a rapid pace (Table 20.1). An example of the rate of change in recommended treatments is the pace of new guidelines by the International AIDS Society–USA panel; they have been revised seven times since 1996. However, while the number of HAART regimens that have been shown to achieve persistently low viral loads continue to increase, they still fall into one of a few broad categories. The original regimens described in 1996 included dual nucleoside therapy accompanied by a PI; now, the nucleoside “backbone” may be supplemented by a non-nucleoside, a “boosted” protease, or a third nucleoside. Currently, there are numerous nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), and PIs approved for therapy, with many others in the pharmaceutical “pipeline.” So, theoretically, a large number of choices within these categories exist. However, certain medications should not be used in combination. For example, ZDV and stavudine (d4T) have overlapping toxicities. Didanosine (ddI) and zalcitabine (ddC) should not be used in combination, and ddI and d4T have been linked to several fatal cases of mitochondrial toxicity in pregnancy.
TABLE 20.1 Abbreviations for Commonly Used Drugs to Treat the Human Immunodeficiency Virus | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 20.2 Recommendations for Initiating Antiretroviral Therapy in Treatment-naive Adults with Chronic Human Immunodeficiency Virus Infectiona | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
The clinician who is preparing to care for an HIV-infected individual must consider the following: when to start therapy, which therapy should be used, and when the regimen should be altered. The trigger for starting antiretroviral therapy in nonpregnant women is either symptomatic HIV disease or, for patients without symptoms, after the CD4 count declines below 350 cells/mm3 but before it reaches 200 cells/mm3 (Table 20.2). Individualization continues to guide the timing of treatment initiation, with consideration of patient readiness, rate of CD4 cell count decline, and plasma HIV-1 RNA level. Thus, with a count between 200 cells/mm3 and 350 cells/mm3, a provider might be more aggressive in promoting the initiation of therapy if the count is dropping rapidly or the viral load is >100,000 copies. In the near future, it can be anticipated that new formulations of antiretroviral drugs and combinations with improved tolerance and convenience may increase the patient and physicians’ willingness to start therapy at an earlier point in the disease.
In regard to the choice of medication with which to begin therapy, several factors need to be considered. Since the number of new drugs continues to multiply, as does information about their benefits and toxicities, it has become increasingly difficult for busy clinicians to stay abreast. There
are several useful resources to assist in that endeavor. The International AIDS Society–USA panel publishes periodic updates to its guidelines. In addition, the Public Health Service (PHS) has established a website that is updated regularly. The expert panel that authors those update guidelines strongly recommends regimens that include either a protease or proteases (indinavir, nelfinavir, ritonavir + saquinavir, ritonavir + indinavir, ritonavir + lopinavir) or a non-nucleoside (efavirenz in the nonpregnant patient) in combination with one of several two NRTI combinations. The USA panel also suggests a combination of two NRTIs with either an NNRTI or a PI boosted with low-dose ritonavir. That panel goes on to state that given the high degree of comparability of the recommended components of these regimens in treatment-naive persons with drug-susceptible virus, the choice of drug centers on acceptability; predicted tolerance; pill burden; comorbid conditions; short-term, mid-term, and long-term adverse event profiles; and successful alternatives in the event the initial regimen fails and drug resistance emerges. The successful outcomes of several “switch studies” suggest that the initial choice of regimen does not preclude safely changing drugs once viral suppression is achieved. There is a great deal of clinical outcome data that support these approaches.
are several useful resources to assist in that endeavor. The International AIDS Society–USA panel publishes periodic updates to its guidelines. In addition, the Public Health Service (PHS) has established a website that is updated regularly. The expert panel that authors those update guidelines strongly recommends regimens that include either a protease or proteases (indinavir, nelfinavir, ritonavir + saquinavir, ritonavir + indinavir, ritonavir + lopinavir) or a non-nucleoside (efavirenz in the nonpregnant patient) in combination with one of several two NRTI combinations. The USA panel also suggests a combination of two NRTIs with either an NNRTI or a PI boosted with low-dose ritonavir. That panel goes on to state that given the high degree of comparability of the recommended components of these regimens in treatment-naive persons with drug-susceptible virus, the choice of drug centers on acceptability; predicted tolerance; pill burden; comorbid conditions; short-term, mid-term, and long-term adverse event profiles; and successful alternatives in the event the initial regimen fails and drug resistance emerges. The successful outcomes of several “switch studies” suggest that the initial choice of regimen does not preclude safely changing drugs once viral suppression is achieved. There is a great deal of clinical outcome data that support these approaches.
A critical factor to recognize is that often there are poor results with antiretroviral regimens when they are used after an initial regimen has failed. This fact suggests that the first regimen affords the best opportunity for long-term control of viral replication. Because the genetic barrier to resistance is greatest with PIs, many would consider a PI with two NRTIs to be the preferred initial regimen. However, efavirenz (an NNRTI) with two NRTIs appears to be at least as effective as a PI with two NRTIs in suppressing plasma viremia and increasing CD4+ T-cell counts, and many would prefer such a regimen initially because it may spare the toxicities of PIs for a considerable time. However, concerns about efavirenz and teratogenicity, that have been demonstrated in animal models as well as by a case report of a myelomeningocele after in utero exposure in a human make this a poor choice for use in early pregnancy. Thus, although the demonstrated ability of efavirenz in combination with two NRTIs to suppress viral replication and increase CD4+ T-cell counts to a similar degree as a PI with two NRTIs supports a preference for efavirenz over other available NNRTIs, in pregnancy the choice of a different NNRTI may be appropriate. Abacavir, a new NRTI, with two other NRTIs (i.e., a triple NRTI regimen) has been used with some success as well. Such a regimen, however, may have short-lived efficacy when the baseline viral load is >100,000 copies/mL. Using two NRTIs alone does not achieve the goal of suppressing viremia to below detectable levels as consistently as does the other regimens discussed previously and should be used only if more potent treatment is not possible. Use of antiretroviral agents as monotherapy is contraindicated, except when there are no other options or in pregnancy to reduce perinatal transmission, as noted below. When initiating antiretroviral therapy, all drugs should be started simultaneously at full dose with the following three exceptions: dose escalation regimens are recommended for ritonavir; nevirapine (NVP); and in some cases, ritonavir plus saquinavir. Recent data confirm that four drugs are generally no better than three drugs when considering treatment with currently available NRTIs and PIs in treatment-naive patients who are not infected with drug-resistant virus.
In order to determine whether a change in therapy is appropriate, the patient must undergo monitoring for immunologic and virologic response as well as for drug toxicity and acceptability. The aim of antiretroviral therapy remains the maintenance of a plasma HIV-1 RNA level below the limits of detection of the most sensitive assays available commercially (i.e., <50 copies/mL). Effective regimens and high levels of adherence result in a decrease of at least 1.0 log10 copies/mL or 90% per month, and suppression of plasma HIV-1 RNA level to below 50 copies/mL will generally be achieved by 16 to 24 weeks, depending on pretreatment level. After antiretroviral therapy is initiated, the plasma HIV-1 RNA level should be checked relatively frequently (e.g., every 4 to 8 weeks) until it is below the limits of detection and regularly thereafter (e.g., three to four times per year). CD4 cell counts generally should be monitored at the same intervals. Resistance testing is recommended in the setting of virologic failure and ideally should be performed when the patient is taking the failing regimen, which maximizes selective pressure on the virus thus increasing the likelihood that resistance testing will detect any mutations that the patient harbors. Genotypic testing for HIV resistance is preferred over phenotypic testing in most settings because it is faster, readily available, and less expensive; phenotypic testing may be more useful for patients with virologic failure following two or more regimens.
There are several reasons to consider modifying a patient’s regimen. One reason would be adverse drug effects. Intolerance or toxicity frequently occurs within the first several weeks of starting a new regimen. In a previously treatment-naive patient who is not expected to harbor archived drug-resistance mutations, if one offending drug can be identified, changing only that drug in an otherwise successful regimen is virologically safe. With some acute toxic effects such as rash, hepatic dysfunction, and febrile systemic reactions, it may be best to stop all antiretroviral drugs. Therapy also may need to be changed because of treatment failure. Treatment failure may be defined virologically, immunologically (declining CD4 cell count), or clinically (HIV-related disease progression). Viral rebound should be confirmed to ensure that it is not transient (i.e., a blip). The fundamental principle for managing any regimen failure, regardless of how many prior regimens the patient has experienced, is to ensure that at least two, and preferably three, drugs used in the new regimen are likely to have activity based on integration of resistance test results and history of antiretroviral regimen use. In individuals in whom the first regimen fails and who were likely infected
with a drug-susceptible virus, a full assessment of adherence is the first step.
with a drug-susceptible virus, a full assessment of adherence is the first step.
Pregnancy
Antiviral medications are used in pregnancy to achieve two goals—to maintain the mother’s health and to prevent mother-to-child transmission (PMTCT) of HIV. In regard to the former, although therapeutic recommendations should not be modified a priori because of pregnancy, a few comments deserve particular mention. The initiation of antiretroviral therapy during the first trimester should be avoided if possible. However, in general, when an HIV-1 infected woman taking effective antiretroviral therapy becomes pregnant, antiretroviral drugs should not be discontinued, though an adjustment in the regimen based on the considerations outlined below may be in order. After the first trimester of pregnancy, the indications for the initiation of therapy are the same as in nonpregnant women with the exceptions noted below. Even if antiretroviral drugs are administered to women for PMTCT of HIV-1, they should be given in combinations intended to be fully suppressive, although some experts will allow for ZDV-only therapy if the viral load is undetectable at the time therapy is initiated (Table 20.3).
Assuming the virus is susceptible, ZDV and lamivudine (3TC) or emtricitabine are the preferred NRTIs. Other NRTIs may be substituted if resistance testing indicates that drug-resistant mutations are present. An increased risk of hepatotoxicity is associated with the use of NVP in pregnancy, especially if initiated in women with >250 CD4 cells/mm3. In women who become pregnant while taking NVP, this risk is substantially lower.
Until more data are available that address concerns about bone formation in utero, tenofovir should be avoided unless resistance testing suggests that its use is advisable. Efavirenz is contraindicated in the first trimester of pregnancy. Nelfinavir has been used extensively in pregnancy, but concerns about its potency make it a less attractive agent. Ritonovir-boosted lopinavir remains an acceptable first-line option, although concerns about dosing have been raised and currently are being addressed.
When possible, ZDV should be used as part of the antiviral regimen. In Pediatric AIDS Clinical Trials Group (PACTG) Protocol 076, which utilized a regimen of ZDV given during pregnancy and labor and to the newborn for 6 weeks, the antenatal dosing of 100 mg administered orally five times daily was selected on the basis of the standard ZDV dosage for adults at the time of the study. However, administration of ZDV three times daily will maintain intracellular ZDV triphosphate at levels comparable with those observed with more frequent dosing. Comparable clinical response also has been observed in some clinical trials among persons receiving ZDV twice daily. Thus, the current standard dosing for ZDV, whether used as a part of a HAART regimen or as single drug therapy for transmission prevention (discussed below), is 200 mg three times daily or 300 mg two times daily. While it is possible that these dosing regimens may not have equivalent efficacy to that observed in PACTG 076, a regimen of two or three times daily is likely to enhance maternal adherence.
Most data regarding the safety of ZDV have been reassuring. Almost 1,000 children have been tracked for 4 years with no increase in risks of neurodevelopmental delay or carcinogenesis. In regard to monitoring of the mother on ZDV, it only is necessary to measure the blood count and liver functions on a monthly basis. The only abnormality that occurs with any frequency is anemia.
Other NRTIs generally have been well tolerated and have not been demonstrated to be teratogenic in humans. However, concerns have been raised about potential adverse effects on both mothers and infants related to the avidity of these drugs for mitochondria. By binding to mitochondrial γ-DNA polymerase and interfering with replication, these drugs can induce mitochondrial dysfunction. ddC demonstrates the greatest inhibition of mitochondrial γ-DNA polymerase, followed in order by ddI, d4T, 3TC, ZDV, and abacavir. Clinical disorders associated with mitochondrial toxicity include neuropathy, myopathy, cardiomyopathy, pancreatitis, hepatic steatosis, and lactic acidosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


