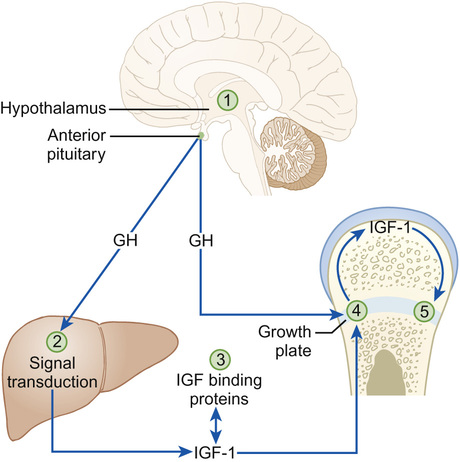
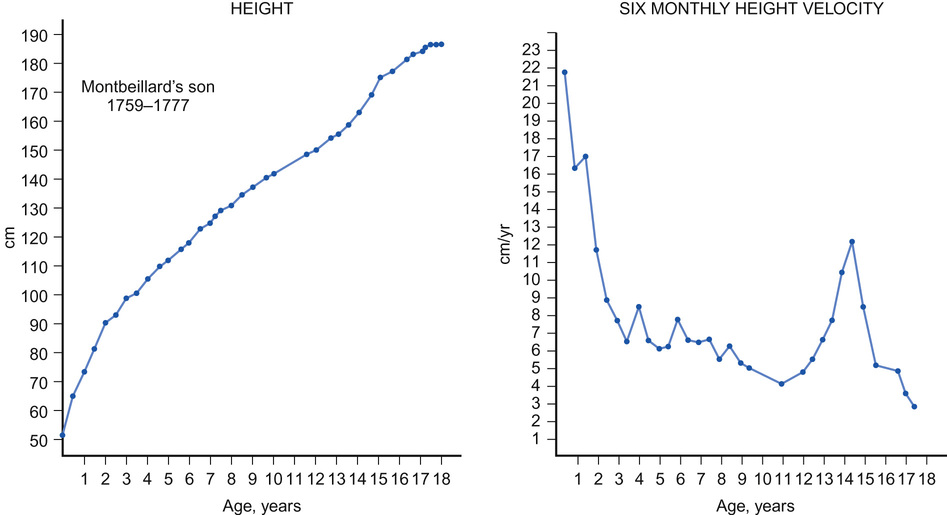
John W Gregory • Understand the basic principles underpinning normal growth • Understand growth measurement and charts and their interpretation • Be able to identify abnormal growth patterns • Know about the genetic and environmental factors that influence growth • Know the common causes and assessment and investigation of short stature • Know the common causes of tall stature • Understand the causes and how to investigate and manage abnormal timing in the onset of puberty The anterior pituitary is derived from the ectoderm, specifically Rathke’s pouch, originally part of palatal development, whereas the posterior pituitary derives from the neuroectoderm. The anterior wall of Rathke’s pouch develops to fill the pouch and form the pars distalis, which forms the bulk of the anterior pituitary, where most hormone production occurs, and the pars tuberalis, which forms a sheath which extends up to the pituitary stalk. The clinical importance of Rathke’s pouch is that it is from this embryonic tissue that a craniopharyngioma is formed. The posterior wall of the pars distalis forms the poorly-defined pars intermedia, which separates the anterior from the posterior pituitary. Mutations of at least eight genes encoding transcription factors involved in pituitary development have been associated with multiple pituitary hormone deficiency. Identification of an underlying genetic defect allows the clinician to anticipate likely pituitary hormone deficiencies that will arise and which will need monitoring. Some of these mutations are listed in Box 12.1. Growth hormone (GH) exerts the major influence on postnatal growth. It is an amino-acid polypeptide, which circulates bound to a GH-binding protein, which is derived from the extracellular component of the GH receptor. GH is secreted from somatotroph cells under the dual regulation of hypothalamically-derived GH-releasing hormone and somatostatin, the combination of which is necessary to produce the episodic pulses, seen mostly overnight and which are essential for normal growth. These hypothalamically-derived regulators of GH secretion are themselves regulated by central neurotransmitters, which integrate input from a variety of stimuli including nutrition, sleep, exercise and stress (Fig. 12.1). GH stimulates the synthesis from the liver and a number of other organs of insulin-like growth factors (IGF-1 and IGF-2), which share a degree of structural homology with proinsulin and may exert weak insulin-like effects. IGF-1 is thought to be a major regulator of GH action. It circulates bound to IGF binding protein-3 (IGFBP-3), whose concentrations are also regulated by GH. This complex associates with another GH-dependent glycoprotein known as acid-labile subunit, the combination forming a ternary complex. Most IGF-1 circulates in bound or inactive forms and the IGF-binding proteins (IGFBPs) prolong the half-life of IGFs, allowing them to be transported to target cells such as in the growth plate, where they interact with IGF receptors to produce their biological effects. We will return to the issue of how IGF-1 can be useful in determining GH abnormalities later in this chapter. The pattern of normal growth can be subdivided into three phases, which correspond to the three main influences on growth. The infancy component, which is largely driven by nutritional factors, depends on normal placental function and nutritional intake in early life. It can be considered to start from conception and it persists through fetal life, ending during the first two years of postnatal life. The childhood phase, which is GH, IGF-1 and thyroxine dependent, starts postnatally and persists until the end of puberty. The final phase is the pubertal component, which is driven by increasing gonadal production of sex steroids, which stimulate GH secretion. These three phases, when superimposed, produce the normal growth pattern (Fig. 12.2) seen through childhood in which height velocity falls rapidly during the first two years of life from approximately 25 cm/year to a fairly steady state of 5–7cm/year through mid-childhood before the onset of the pubertal growth spurt. In girls, the pubertal growth spurt starts at the onset of breast development, peaking a year later at an average age of 12 years, whereas in boys the growth spurt is typically two years later, peak height velocity coincident with testicular volumes of 15 mL. The average difference of 12.5–14 cm in the adult height of men and women is caused by the longer prepubertal contribution to growth and greater peak height velocity seen in boys. Growth failure can arise from genetic abnormalities, nutritional and endocrine problems and defects in almost any organ system. Therefore, when assessing a child referred with a possible growth disorder, a detailed and wide-ranging history is required. Details should be sought of: • Pregnancy, mode of delivery and birth weight, which may impact on the infant phase of growth • Development of signs of puberty • Headache, visual disturbance or symptoms to suggest pituitary dysfunction or intracranial disease • Details of systemic symptoms that might suggest any coexistent medical disorder • Social history, including details of how the short stature is affecting the child. In a child with unusually tall stature, in addition to the family history, specific enquiry should be made for: A fundamental requirement when examining children with growth disorders is an accurate measurement of their growth. Because of the effect of time of day (human height shortens as the day progresses) and inter-observer variation on measurement, serial growth measurements should be undertaken at approximately the same time of day and preferably by the same measurer. Over the age of two years, a stadiometer (preferably wall-mounted) should be used and the child measured without shoes or socks, with heels, buttocks and shoulders against the backplate and the head in the Frankfurt plane (an imaginary line connecting the lower border of the eye socket with the external auditory meatus). There is no evidence that undertaking stretched measurements with upward pressure on the mastoid processes achieves greater consistency, but the same technique should be used when comparing sequential measures. Under the age of two years, supine table measurements or a neonatometer and two observers are required to assess length, ensuring that the Frankfurt plane is vertical and that the child’s head is in firm contact with the headboard and the foot dorsiflexed against the movable baseplate. As skeletal dysplasias may impair growth of different parts of the skeleton differentially, the sitting height, which is a proxy for vertebral body growth, should be measured using a table-mounted stadiometer. Subtracting sitting height from standing height produces the sub-ischial leg length, which is a measure of long bone growth in the leg. Head circumference should be measured in children under the age of two years. Height measurements should be compared with weight (assessed in a child wearing minimal clothing) by plotting measurements on a growth chart. In the UK, the UK-WHO growth charts are used. These are a composite of the UK90 charts derived from cross-sectional growth data and the WHO growth standards, which describe the growth of healthy breastfed children from six countries. Because of the association of weight with height (taller children tend to be heavier), interpretation of weight data requires adjustment for height. This should be done by calculating the body mass index (weight in kilograms / (height in metres)2) and plotting this on a centile chart. To help interpret the growth pattern, height measurements should be compared with other measurements taken in the past by plotting all available measurements on a growth chart to evaluate whether crossing of centiles has occurred, which implies an abnormal height velocity. The appropriateness of the child’s height for their genetic background is assessed by calculating the target height range (requires parental height measurements), which is the mid-parental centile (the mid-point between the parents’ centiles) +/− 8.5 cm. Additional features that should be assessed on thorough physical examination of a child with abnormal growth include: Before considering any investigations, the findings from the history and examination should be integrated into a differential diagnosis. The following investigations should be considered: • In a short or slowly-growing child with no obvious pathology, an X-ray of the left wrist to calculate a bone age should be performed to assess the degree of delay in physical development, along with a blood sample for: – Full blood count, blood film and ESR or C-reactive protein – Urea, electrolytes and creatinine – Karyotype in girls to exclude Turner’s syndrome In a tall or rapidly-growing child, the following investigations may be necessary: • Bone age, which is useful to assess physical maturation • Karyotype in boys to exclude Klinefelter’s syndrome • Thyroid function tests for hyperthyroidism • IGF-1 to exclude GH oversecretion • Cardiac ultrasound if Marfan’s syndrome is suspected • FMR1 gene analysis to exclude Fragile X syndrome in a boy with learning difficulties • DNA for specific genetic syndromes (e.g. Marfan’s, Beckwith–Wiedemann, Sotos syndromes). Familial short stature is the most common cause of short stature referred to clinical services. Calculating the target height range is required to demonstrate familial short stature, though if one parent is particularly short, one needs to consider whether they too may have a potentially inheritable underlying growth disorder. Children with familial short stature will have a height centile consistent with their short target height range and will demonstrate growth parallel to the centiles and consistent with a normal height velocity. Extensive investigations are not indicated but a careful explanation and reassurance for the child and parents is required to alleviate anxiety. Short stature due to constitutionally delayed puberty is discussed in the section on puberty. Short stature due to being small for gestational age is suggested when the birth weight is below the 10th centile. Symmetrical growth failure implies adverse influences on fetal growth that have operated throughout much of development, whereas asymmetric growth failure in which head circumference is preserved implies growth failure restricted to the last part of pregnancy, often due to placental failure. There are many other underlying causes including: Most (80–85%) small-for-gestational-age infants will show catch-up growth postnatally but those with symmetrical growth failure are least likely to do so. Infants who are small for gestational age are at increased risk of hypoglycaemia in the first two days of life. In the much longer term, they have been shown to be at increased risk of a range of adult diseases, such as hypertension, cardiovascular disease, type 2 diabetes and obesity. A fetal programming hypothesis (the Barker hypothesis) has been proposed, which suggests that there are critical periods in fetal development when permanent adaptive changes occur in body structure and function to cope with nutritional insufficiency in utero and likely nutritional challenges postnatally. However, these adaptations place the individual at a disadvantage postnatally should calorie supplies become potentially unlimited. Treatment of affected individuals involves ensuring an adequate supply of nutrition postnatally to facilitate catch-up growth. In those individuals who remain short, GH therapy at larger doses than those required to treat GH deficiency has been shown to increase final height, though whether this impacts on other risk factors for disease in later life is unknown. The more common syndromic causes of short stature which may present with growth concerns include Russell–Silver syndrome, fetal alcohol spectrum disorder, Turner’s syndrome and Noonan’s syndrome. Russell–Silver syndrome is an example of an imprinted disorder, usually caused by maternal uniparental disomy of chromosome 7. Imprinted disorders are associated with assisted reproductive techniques and often cause abnormalities of growth. Russell–Silver syndrome is characterized by intrauterine growth restriction, increased risks of hypoglycaemia and sweating, asymmetry (one side of the body being shorter than the other) and short stature with failure to catch up growth, thinness, a triangular-shaped face with a small pointed chin and clinodactyly. Treatment involves optimal dietary support and, for some, GH therapy. Alcohol is a teratogen and exposure in utero causes a number of problems, including impaired growth at any time point postnatally, brain damage leading to poor concentration, behaviour and learning difficulties and a characteristic facial appearance of microcephaly, a flat mid-face, low-set ears and micrognathia. This is caused by a loss or abnormality of one X chromosome, affecting 1 in 2500 girls. Although the phenotype is relatively mild for such a major chromosomal anomaly, reflecting the partial inactivation of the second X chromosome from early fetal life, 99% of conceptions result in miscarriage or stillbirth. About one third of genes on the short arm (Xp) are unsilenced, including the short stature homeobox (SHOX) gene, and these account for the characteristic features, which include: a skeletal dysplasia causing short stature; short fourth and fifth metacarpals; cubitus valgus; micrognathia; ovarian failure leading to pubertal failure and infertility; lymphoedema; neck webbing; a low hairline and increased naevi; congenital heart disease, particularly coarctation of the aorta; wide-spaced nipples; Madelung deformity (a focal dysplasia of the distal radial physis), middle ear problems; renal anomalies; specific learning difficulties related to numeracy and visuospatial tasks; social vulnerability; and an increased risk of autoimmune and inflammatory disease. Many children with Turner’s syndrome have few abnormal findings and so karyotyping is essential in any girl with impaired growth of unknown aetiology. Treatment requires GH therapy to improve growth, oestrogen induction of puberty and specific monitoring for cardiac, renal, autoimmune and hearing abnormalities. Noonan’s syndrome may affect as many as 1 in 1000 individuals. It is caused by mutations of genes involved in the RAS/MAPK signalling pathway (including the PTPN11, SOS1, KRAS and RAF-1 genes) and is inherited in an autosomal dominant manner. Clinical features include short stature, scoliosis, low-set ears, ptosis, pectus excavatum, cubitus valgus, pulmonary stenosis, cryptorchidism and delayed puberty, lymphoedema, mild educational difficulties and a coagulation defect. A range of bony dysplasias may cause short stature. Depending on which part of the skeleton is involved, impacts on growth may lead to skeletal disproportion, which justifies the measurement and comparison of both sitting and standing heights. Many skeletal dysplasias are inherited in an autosomal dominant pattern and so the possibility of an affected parent of a short child should be considered. Achondroplasia and hypochondroplasia cause rhizomelic (shortening of the proximal limb segment) short stature and are often found to be caused by mutations of the fibroblast growth factor receptor 3 (FGFR3) gene. The mutated receptor is constitutionally active and inhibits cartilage formation and thus bone growth. Spondyloepiphyseal dysplasia leads to markedly impaired trunk growth and less severely affected short limbs, causing a disproportionately short sitting height. In children with short-limbed forms of short stature, height may be improved by leg-lengthening surgery. Chronic disease of many systems may lead to impaired growth. Mechanisms involved include a negative energy balance due to:
Growth and puberty
Normal growth
Embryology of the pituitary
Endocrine regulation of growth
Physiology
History
Examination
Investigations
Short stature and impaired growth
Familial short stature and constitutionally delayed growth and puberty
Small for gestational age
Syndromic short stature
Russell–Silver syndrome
Fetal alcohol spectrum disorder
Turner’s syndrome
Noonan’s syndrome
Skeletal dysplasias
Chronic disease
Growth and puberty
Chapter 12
Learning objectives
By the end of this chapter the reader should: