Daniel Carroll, Charlotte Slaney With contributions by, John W Gregory
Genital disorders
Genital anomalies in children are common, and are the cause of much anxiety for children and their parents. The first question often asked of new parents after delivery is what sex their baby is, and this clearly demonstrates the importance within our society of normal genitalia and sexual identity. The development of the genitalia is complex, and as there are multiple steps in development there are many opportunities for congenital or acquired abnormalities to develop. Although most are detected at birth, they are increasingly recognized antenatally. However, congenital problems, particularly in the development of the internal genitalia, may not be detected until later in life.
An understanding of the normal embryology of sexual differentiation is fundamental to understanding urogenital abnormalities.
Genetic aspects of genital development
Genetic sex is established at conception by the chromosome complement of the egg (23 X) and the sperm that fertilizes it (23 X or 23 Y). The presence of at least one X chromosome appears to be necessary for the survival of individual cells.
Major chromosomal abnormalities can occur during gametogenesis, or during the first few cell divisions (which results in mosaicism). Failure of sex chromosomes to separate fully during gametogenesis may result in Turner’s syndrome (45 XO) and Klinefelter’s syndrome (47 XXY).
Abnormalities of the sex chromosomes may occur in two distinct cell lines derived from the same zygote (sex chromosome mosaicism). The genetic imbalance arising from chromosomal abnormalities is expressed as profound disturbances of embryogenesis across a number of systems, including the genitourinary tract.
The majority of congenital anomalies do not have such a clearly defined genetic basis. Most are either sporadic or, if genetic, have variable expression and penetrance and are likely to involve the interaction of multiple genes rather than a single gene mutation.
Embryology of the genital tracts
The internal and external genitalia of both sexes are genetically programmed to differentiate as female. The presence of an active SRY gene on the Y chromosome causes development to move towards the male phenotype (Fig. 20.1). Until the 6th week of gestation, the embryonic precursors of the genitalia of both genetically male and genetically female embryos share identical embryonic precursors.
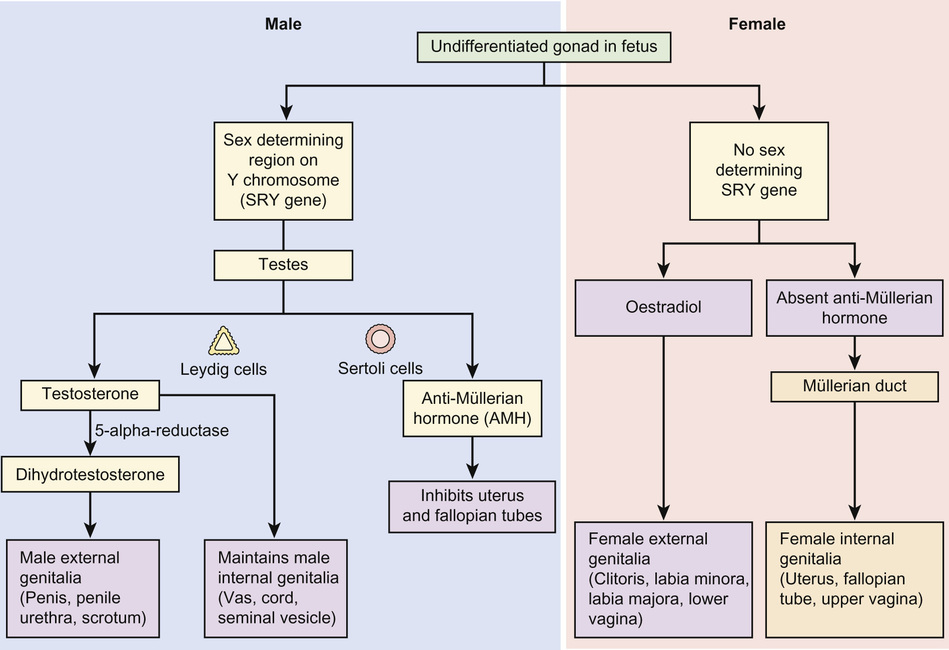
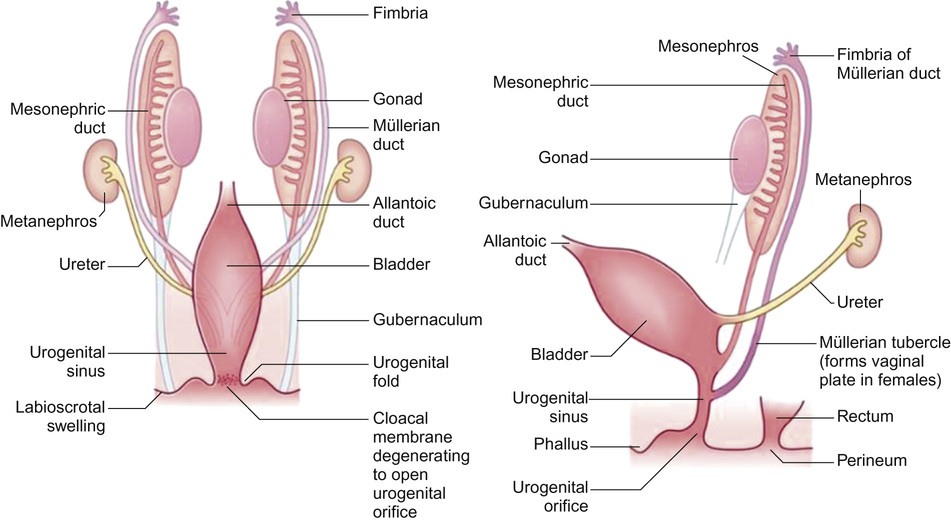
Differentiation of the gonads and the genital tracts is initiated by the migration of primordial germ cells from the yolk sac to the lumbar regions of the embryo. This occurs at around the 6th week of gestation (Fig. 20.2). The germ cells and the surrounding mesenchyme coalesce to form the primitive sex cords. At the same time the paired paramesonephric ducts appear. From this stage onwards, the pathways of development of the genetically male and the genetically female embryo begin to diverge as sexual differentiation begins to be phenotypically expressed.
Female development
Internal genitalia
The primitive sex cords degenerate and secondary sex cords develop from the mesoderm of the genital ridge enfolding the primordial germ cells to form primitive ovarian follicles. Differentiation of the genitalia down a female pathway is not entirely passive, as the normal development of the ovary requires two normal X chromosomes (females with Turner’s syndrome (XO) typically have streak ovaries). In the absence of testosterone, the mesonephric ducts regress leaving only vestigial remnants. The paramesonephric ducts persist as the fallopian tubes. The fused distal portions of the paramesonephric ducts give rise to the uterus and upper two thirds of the vagina (Fig. 20.3).
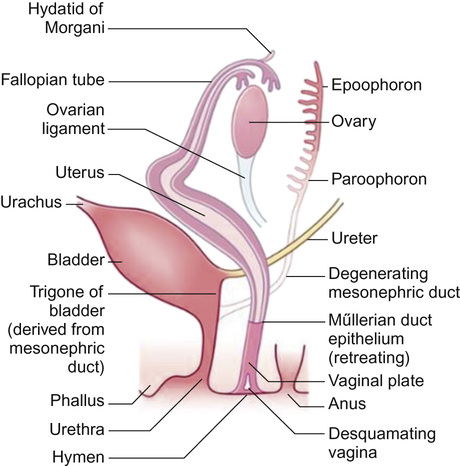
Distally, the paired paramesonephric ducts fuse with the urogenital sinus. Between the 10th and 20th week of gestation, displacement of the sinuvaginal bulb towards the perineum separates the opening of the vagina from the perineum. The upper two thirds of the vagina forms from the paramesonephric ducts, whereas the distal third has its origins in the urogenital sinus. The introitus and external genitalia are derived from ectoderm.
External genitalia
In the absence of androgens or effective androgen receptors, the external genitalia progress down a pathway of female development. The genital tubercle becomes the clitoris, the urogenital sinus becomes the vestibule of the vagina, the urogenital folds persist as the labia minora and the labioscrotal folds persist as the labia majora.
Male development
The differentiation of the development of the male sexual phenotype is initiated by the SRY gene located on the Y chromosome, mediated by multiple other Y chromosome and autosomal ‘downstream’ genes. The gene products expressed by the SRY gene stimulate the medullary sex cords to differentiate into secretory pre-Sertoli cells. From the seventh week of gestation, these cells secrete anti-Müllerian hormone (AMH) – a glycoprotein that plays a central role in subsequent differentiation of the male genital tract.
In the male, the paramesonephric ducts disappear leaving behind only the vestigial remnants (the appendix testis and utriculus). From the seventh week of gestation, the urogenital sinus advances onto the developing phallus as the urethral groove. Ingrowth of the urethral groove is associated with the appearance of urethral plate tissue, which subsequently canalizes to form the anterior urethra (Fig. 20.4). Closure of the urethra should be complete from around 15 weeks’ gestation.
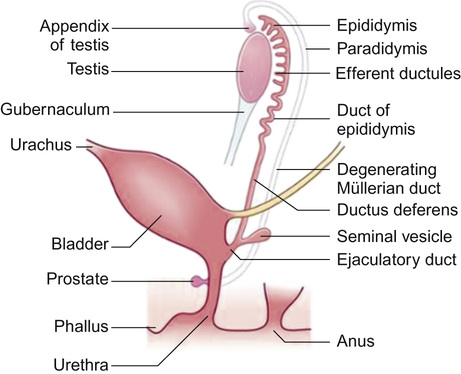
The testis
Testicular descent occurs in two distinct phases. The first phase (abdominal phase) is initiated by the presence of anti-Müllerian hormone (AMH). Thus, the testis may descend to the internal ring from the lumbar region without active Leydig cells.
The gubernaculum (Latin for rudder) extends down to the labioscrotal folds and a second more active phase of testicular descent begins between 25 and 30 weeks’ gestation. Under the influence of testosterone, the gubernaculum contracts, pulling the testis into its definitive scrotal position. Abnormal positioning of the gubernaculum leads to ectopically positioned testes; a failure in testosterone production or testosterone recognition results in non-descent or incomplete descent. In summary:
In response to testosterone, the mesonephric (Wolffian) duct differentiates between the 8th and 12th week of gestation, giving rise to the epididymis, rete testes, vas deferens, ejaculatory ducts and seminal vesicles.
External genitalia
The complete and adequate differentiation of the male external genitalia is dependent upon a number of different factors. Firstly, the production of testosterone by the primitive Leydig cells of the embryonic testis. This is converted into the more physiologically active dihydrotestosterone by the enzyme 5α-reductase. Active androgen receptors within target cells are required to complete the androgenic stimulation of the male external genitalia. Under the influence of dihydrotestosterone, the genital tubercle develops into the male phallus.
Male genitalia
At birth, it is straightforward to identify a phenotypically normal male infant. Abnormalities of the external genitalia are seen in around 2% of male infants at birth. The penis should be straight and sit cranial to the scrotum, the prepuce should be completely formed (although there are a wide variation of normal variants), and the external urethral meatus should be hidden behind the prepuce and located centrally at the tip of the penis (i.e. distal). It is painful to retract the prepuce as male infants are typically born with a physiological phimosis and so retraction to visualize the external urethral meatus should not be attempted. If the urethral meatus and glans is visible at birth, it should prompt more detailed examination, as an incomplete or hooded foreskin is almost always present in infants with hypospadias.
In the term male infant, the testes should be present bilaterally and be easily drawn to the base of the scrotum. The scrotum should be well-developed to accommodate the testes. In the prepubertal male, the size of the testis should be roughly the same size as the glans penis and symmetrical. The stretched length of the penis from the pubic tubercle should be greater than 2.5 cm. A penis smaller than this should prompt more detailed examination and investigation for ambiguous genitalia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


