Genetics in Obstetrics and Gynecology
Kenneth Ward
Stating that “the rapid growth and clinical adaptation of genetically based information and technology are fundamentally changing the practice of medicine in general, and obstetrics and gynecology in particular,” the Council on Resident Education in Obstetrics and Gynecology added a seventh unit to the residency curriculum—a unit on genomics. The two fundamental paradigms in biology are that all health and disease have a molecular basis based on the deoxyribonucleic acid (DNA) “blueprint” and that phenotypes evolve through changes in the genome. Thus, genomics—the study of genes and their functions—has arguably become the central science of medicine.
Genetic variables affect most diseases. Mutations may cause or serve as a cofactor for disease; polymorphisms may protect against disease or affect responses to treatment. Genetic screening has become an increasingly important component of prenatal care. Recent discoveries have expanded the indications for cytogenetic or molecular genetic tests performed on chorionic villi, amniocytes, and fetal blood. First-trimester DNA-based prenatal diagnosis is now possible for hundreds of conditions. New technologies enable testing of a single cell for chromosomal or mendelian problems, allowing preimplantation testing of embryos, and minimally invasive screening using the small population of fetal cells, which are present in the maternal circulation. Gene therapy is being applied, even prenatally, to treat inborn errors and genetic diseases. The expansion of preventative and therapeutic options will provide additional impetus for genetic evaluation.
More than 25 million Americans have a diagnosed genetic disease. One percent of all newborns have a recognizable mendelian disorder; 0.5 percent have a chromosomal syndrome; and many more have a polygenic, multifactorial disorder. Most pregnancy losses and most congenital anomalies have genetic causes. Genes also play an important role in common gynecologic disorders such as leiomyomata, endometriosis, gynecologic cancers, and infertility. There are inherited tendencies to have multiple gestations, preeclampsia, gestational diabetes, and other pregnancy complications. Susceptibilities to infectious or teratogenic agents are usually genetically determined.
Recognizing that a condition is genetic enables us to find the gene responsible for the trait or illness, which can lead to improved means of, classification, diagnosis, prevention, and treatment. Correlation of the phenotype with the genotype often provides specific predictive insights. Because any DNA test can be performed prenatally, discovery of the gene that causes a particular disease can give at-risk couples the necessary information to prepare for having an affected child, to consider prenatal therapy if available, or to choose to terminate the pregnancy.
Patients often have genetic illnesses that will affect pregnancy or their gynecologic care, illnesses that may remain undiagnosed unless physicians are thorough in their evaluation of unusual signs or symptoms. For example, a new obstetric patient presents with a wasted facial appearance, generalized weakness, and difficulty releasing her grip when shaking the physician’s hand. Hopefully, this patient has already been evaluated and correctly diagnosed. If not, it is the obstetrician’s responsibility to refer a patient with unusual signs and symptoms for evaluation. In this case, the patient has the classic signs of myotonic dystrophy, an autosomal dominant disorder. Formerly, myotonic dystrophy was a difficult diagnosis to establish in some patients, but the diagnosis can be made easily by using DNA analysis. The physician and this woman need to know that she is at significant risk for developing polyhydramnios, which could result in preterm labor; her fetus may have positional
deformities and is at risk for a severe, often fatal, neonatal form of myotonia; her labor is likely to be prolonged, and she may be unable to push in the second stage. If she requires a cesarean section, she may have undiagnosed cardiac problems, which place her at higher risk for anesthesia. If failure to recognize these risks results in a bad outcome, a lawsuit claiming negligence might be brought.
deformities and is at risk for a severe, often fatal, neonatal form of myotonia; her labor is likely to be prolonged, and she may be unable to push in the second stage. If she requires a cesarean section, she may have undiagnosed cardiac problems, which place her at higher risk for anesthesia. If failure to recognize these risks results in a bad outcome, a lawsuit claiming negligence might be brought.
Patterns of Inheritance
Single Gene (Mendelian) Disorders
The observations that “like begets like” has been stated throughout recorded history, but current theories describing how genetic traits and illnesses are inherited are just over 100 years old. In the late 1800s, Mendel described how individual genetic traits were passed on from generation to generation. Single gene disorders (i.e., mendelian disorders) are conditions caused by a mutation at a single site in the DNA and inherited in the proportions predicted by Mendel’s laws. These disorders can be dominant conditions in which the phenotype is expressed even when only one chromosome of a pair has a defect or recessive conditions that are only expressed if the defect exists on both chromosomes. Classically, these different modes of inheritance are revealed by pedigree analysis (Figs. 6.1, 6.2).
As more information about the tremendous variation that occurs at every locus becomes available, the distinctions between dominant and recessive conditions have become blurred. Dominance and recessiveness are attributes of the phenotype, not attributes of the gene or allele. They are empiric terms, and they depend on the sensitivity of the method used to describe the phenotype. The researchers must specify particular phenotypic features when describing inheritance. For instance, sickle cell anemia is a recessive disease only if the full-blown disease is considered, but it is a codominant condition if the hemoglobin is being analyzed by electrophoresis. The ABO blood group is an example of a trait in which both codominant and recessive inheritance is seen. The retinoblastoma gene is a recessive tumor suppressor gene at the cellular level, but abnormalities in the gene are responsible for the autosomal dominant tendency to develop retinoblastomas and osteosarcomas.
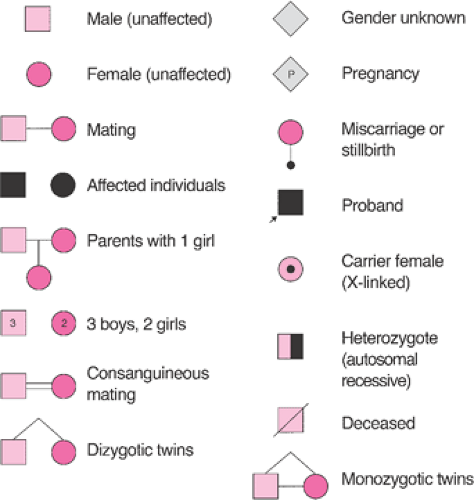 Figure 6.1 Symbols used to draw a pedigree. |
In autosomal dominant conditions, the disease is expressed in persons who are heterozygous for the disease-causing mutation. McKusick’s catalog of mendelian disorders describes more than 3,000 dominant conditions. Marfan syndrome, myotonic dystrophy, neurofibromatosis, achondroplasia, and Huntington disease are examples of autosomal dominant disorders. The probability of an affected person transmitting the abnormal gene to his or her children is 50% with each pregnancy. Typically, autosomal dominant conditions have less than 100% penetrance, and fewer than 50% of the offspring show signs of the disorder. Male and female offspring are usually affected with equal frequency and severity. The trait passes through one parental line only, and father to son transmission can occur. For highly penetrant, autosomal dominant conditions, the gene is expressed in each generation (i.e., vertical transmission). New mutations are relatively common, and on average, paternal age is advanced when isolated, sporadic, or new mutation cases appear. Autosomal dominant phenotypes often involve isolated or multiple structural defects. They can be extremely variable, and the onset of clinical features is often age dependent. Dominant disorders tend to be less severe than recessive diseases, but they are usually lethal in the rare persons who are homozygous for a dominant disease.
Autosomal recessive conditions are only expressed in persons in whom both versions (i.e., alleles) of the involved gene are abnormal. More than 1,500 autosomal recessive conditions have been described. Cystic fibrosis, sickle cell anemia, Tay–Sachs disease, and phenylketonuria are examples of autosomal recessive disorders. Male and female offspring are affected with equal frequency and severity. Each parent is a heterozygous carrier, and abnormal genes are inherited from both parents. Each offspring of two carrier parents has a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of being neither a carrier nor affected. If the recessive phenotype is extremely rare, consanguinity is usually found in the pedigree. Affected persons rarely have affected children; autosomal recessive inheritance shows a “horizontal” pattern in a pedigree, with typically only a single generation of siblings affected. Affected persons who mate with unaffected persons who are not carriers have only unaffected, carrier
offspring. Most autosomal recessive phenotypes are biochemical or enzymatic in nature, and they tend to be less variable and more severe than dominant conditions.
offspring. Most autosomal recessive phenotypes are biochemical or enzymatic in nature, and they tend to be less variable and more severe than dominant conditions.
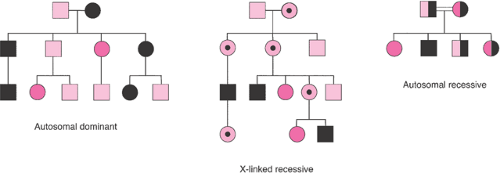 Figure 6.2 Patterns of inheritance revealed in pedigree analysis. |
X-linked inheritance occurs when a trait is carried on the X chromosome. Boys are hemizygous for X chromosome genes, but girls can be homozygous or heterozygous. Of the 300 X-linked recessive diseases that are recognized, the hemophilias and Duchenne muscular dystrophy are the best known. Characteristics of X-linked recessive inheritance include a higher incidence of the disorder in male than female offspring. The mutant gene or disease is never transmitted directly from father to son, and all the daughters of an affected man are carriers. The trait is transmitted through carrier females, and affected males in the same kindred are related to one another through the females. X-linked dominant diseases are much rarer; examples include Alport syndrome, vitamin D resistant rickets, and incontinentia pigmenti. They appear twice as often in female as in male offspring. All daughters of an affected man have the disorder, but no sons are affected. Heterozygous affected women transmit the mutant allele at a rate of 50% to progeny of both sexes. If the affected woman is homozygous, all of her children will be affected.
Y-linked or holandric inheritance occurs when a trait is carried on the Y chromosome. Only male offspring are affected, and there is only male-to-male transmission. There are no known disease genes that are inherited in this fashion, but genes for gender determination, tooth size, and height occur on the Y chromosome.
Mitochondrial inheritance is also relatively uncommon; these traits and disorders are inherited through mutations on the mitochondrial chromosome (Fig. 6.3). Since mitochondria are inherited exclusively with the cytoplasm of the egg, a woman who carries a disease will pass the disease to 100% of her offspring. Male carriers will pass the disorder to none of their offspring. Mitochondrial diseases often affect tissues dependent on the chemical energy produced by mitochondria; Leber optic atrophy and certain rare myopathies are examples.
Polygenic, Multifactorial Disorders
Multifactorial or polygenic inheritance is the most common form of inheritance. Even in the classic mendelian disorders described previously, there can be tremendous quantitative and qualitative differences in the phenotype between persons who have the same allele or the same genetic mutation. This variability can be evident as nonpenetrance of certain features (or the entire phenotype) and as differences in the severity of features, the frequency of cyclic or episodic events, or the age of onset of the first clinical sign of the disorder. The underlying genetic background of the affected person, including gender influences and limitations, can cause genetic variability. The phenotype may be further influenced by maternal factors such as cytoplasmic inheritance, the intrauterine environment, or imprinting. X-linked disorders can be altered by variations in X inactivation or lyonization. Each genotype undergoes subtle changes through somatic mutation, gene amplification, or transpositions and positional effects over time. Exogenous factors such as the environment, teratogens, medical intervention, and chance also influence variability.
Most congenital anomalies show multifactorial inheritance (Table 6.1). A common error made by some obstetricians is to counsel a patient that rare conditions will not occur repetitively in her family. If the birth defect in question has a strong genetic component or if there is an identifiable environmental or teratogenic component, which would recur, in subsequent pregnancy, the risks may remain high for that patient (Table 6.2). The rates may be even higher if a mendelian or chromosomal condition has gone unrecognized in the affected child. Before counseling patients about the recurrence risk of any birth defect, it is pertinent to review which syndromes are associated with that birth defect and ask whether any member of the family has those syndromes. When this requires skill or knowledge beyond the usual expertise of an obstetrician-gynecologist, referral to a medical geneticist is appropriate.
Multifactorial inheritance usually works according to a threshold model (Fig. 6.4). Several factors must
collaborate to cause a bodily function to go awry, and only after these factors reach some critical point is the phenotypic effect seen. Many different factors can affect the observed recurrence risk. The intrinsic heritability “or geneticness” of the condition is frequently the most important factor. This is usually determined by examining whether monozygotic twins are concordant for a particular condition relative to dizygotic twins. For instance, neonatal seizures show a very high heritability rate, with 85% to 90% concordance in monozygotic twins versus 10% to 15% concordance in dizygotic twins. The population incidence of the condition is another important variable. The recurrence risk is higher for common disorders or within populations with a high incidence of the disorder. In disorders with a relatively high heritability, the recurrence risk of the disorder approximates the square root of the population incidence. There can be marked variation in the population frequency of different disorders and different ethnic groups. For instance, cleft lip occurs commonly in Native Americans, but African Americans have a ninefold lower incidence of cleft lip than the general population.
collaborate to cause a bodily function to go awry, and only after these factors reach some critical point is the phenotypic effect seen. Many different factors can affect the observed recurrence risk. The intrinsic heritability “or geneticness” of the condition is frequently the most important factor. This is usually determined by examining whether monozygotic twins are concordant for a particular condition relative to dizygotic twins. For instance, neonatal seizures show a very high heritability rate, with 85% to 90% concordance in monozygotic twins versus 10% to 15% concordance in dizygotic twins. The population incidence of the condition is another important variable. The recurrence risk is higher for common disorders or within populations with a high incidence of the disorder. In disorders with a relatively high heritability, the recurrence risk of the disorder approximates the square root of the population incidence. There can be marked variation in the population frequency of different disorders and different ethnic groups. For instance, cleft lip occurs commonly in Native Americans, but African Americans have a ninefold lower incidence of cleft lip than the general population.
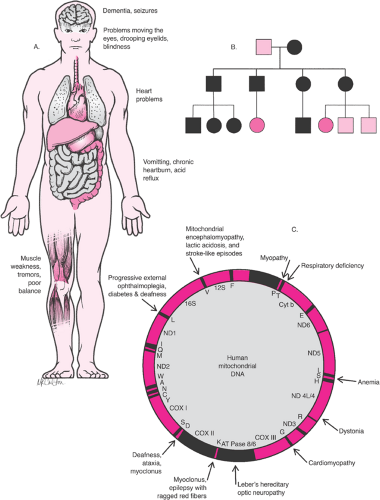 Figure 6.3 Mitochondrial diseases. A: Features of mitochondrial diseases. B: Mitochondrial (cytoplasmic) pedigree analysis showing pattern of inheritance. C: Disease map of mitochondrial DNA. |
TABLE 6.1 Types of Birth Defects | |
|---|---|
|
TABLE 6.2 Empiric Recurrence Risks for Common Congenital Anomalies | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
If the incidence of a congenital anomaly shows a sex bias, the recurrence risk is higher in the offspring (and other relatives) if the parent is the less frequently affected sex. For example, pyloric stenosis affects five times as many male as female offspring, and empiric data show that there is a 25% chance of producing an affected child if the mother
had pyloric stenosis at birth and only a 4% risk if the father is the affected parent. Similarly, the recurrence risk is higher when the gender of the affected child is the less frequently affected sex. Again, the risk of having another child with pyloric stenosis is 3.2% if the first affected sibling is a boy and 6.5% if the affected sibling is a girl. Hirschsprung disease, clubfoot, and cleft lip are examples of anomalies that are more common in male infants: cleft palate, anencephaly, hip dysplasia, and scoliosis are more common in female infants.
had pyloric stenosis at birth and only a 4% risk if the father is the affected parent. Similarly, the recurrence risk is higher when the gender of the affected child is the less frequently affected sex. Again, the risk of having another child with pyloric stenosis is 3.2% if the first affected sibling is a boy and 6.5% if the affected sibling is a girl. Hirschsprung disease, clubfoot, and cleft lip are examples of anomalies that are more common in male infants: cleft palate, anencephaly, hip dysplasia, and scoliosis are more common in female infants.
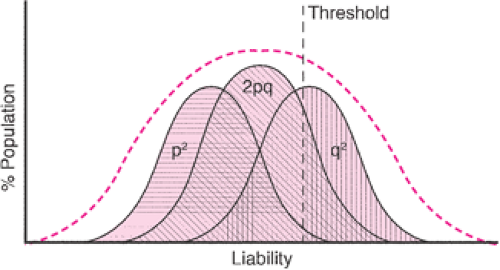 Figure 6.4 Polygenic multifactorial inheritance. Individuals homozygous for the p allele are at decreased risk; while individuals homozygous for the q allele are at increased risk for developing the disease. Heterozygous individuals have an intermediate risk. |
The number of affected individuals in a kindred can affect the recurrence risk. The greater the number of family members who have already been affected with a multifactorial condition, the more likely it is that the genetic background is favorable for expression of this condition. After a couple has one affected child with cleft lip and palate, there is a 4% empiric recurrence risk. After two affected children, the risk rises to 10%. Consanguinity also increases the risk of recurrence because of the greater likelihood of deleterious genes being shared, but a more distant relationship from an affected person decreases the risk of recurrence.
The severity of the disorder often predicts the recurrence risk. An illustration of this is found in Hirschsprung disease; the recurrence risk is proportional to the length of the aganglionic segment of the colon. Neural tube defects are the most notable exception to this rule, because the recurrence risk for any neural tube defect appears to be the same whether the first affected child had anencephaly or a small spina bifida lesion.
Cytogenetic Disorders
Cytogenetic disorders are changes in the genome visible under light microscope. These gross lesions involve the loss or duplication of a large number of genes; multiple malformations and dysfunctions are usually observed clinically. Diagnostic clues for a cytogenetic disorder can range from subtle dysmorphic features to major structural malformations, particularly craniofacial, skeletal, cardiac, and genitourinary malformations. No individual anomaly is pathognomonic for a particular chromosomal syndrome; rather, it is the pattern that can be distinctive. There is tremendous overlap between patterns, and because nonchromosomal syndromes can mimic chromosomal abnormalities, obtaining a karyotype is always necessary to confirm the diagnosis. Cytogenetic disorders are usually associated with some degree of mental retardation and growth deficiency. Most have an increased rate of perinatal loss and premature mortality of live-born neonates. The rate of chromosome abnormalities is at least 40% to 60% in first-trimester abortuses, and the rates of abnormalities are also elevated in fetal deaths and preterm and post-term deliveries (Tables 6.3, 6.4). About 1 in 160 babies is born with a genetic defect detectable by ordinary cytogenetic means (Table 6.5).
TABLE 6.3 Incidence of Chromosomal Aberrations in Pregnancy Losses at Various Gestational Ages | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Cytogenetic studies have been used clinically for approximately 40 years. In the late 1950s, it was determined that humans have 46 chromosomes and that many of the recognized birth defect syndromes, such as Down
syndrome, Turner syndrome, and Klinefelter syndrome, have abnormalities of chromosome number or structure. Normally, the nucleus of most human cells contains two sets of chromosomes, with one set contributed by each parent. Each set has 22 autosomes and either an X or a Y sex chromosome (Fig. 6.5). Cells duplicate their chromosomes through mitosis; germ cells divide by a related process called meiosis (Fig. 6.6). Errors in meiosis or mitosis can cause the resulting cells to have an incorrect number of chromosomes, called aneuploidy (Fig. 6.7).
syndrome, Turner syndrome, and Klinefelter syndrome, have abnormalities of chromosome number or structure. Normally, the nucleus of most human cells contains two sets of chromosomes, with one set contributed by each parent. Each set has 22 autosomes and either an X or a Y sex chromosome (Fig. 6.5). Cells duplicate their chromosomes through mitosis; germ cells divide by a related process called meiosis (Fig. 6.6). Errors in meiosis or mitosis can cause the resulting cells to have an incorrect number of chromosomes, called aneuploidy (Fig. 6.7).
TABLE 6.4 Types of Chromosomal Abnormalities in Spontaneous Abortuses | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 6.5 Incidence of Chromosomal Aberrations Seen in Newborn Surveys | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
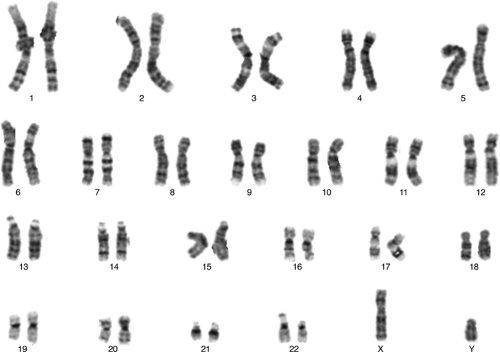 Figure 6.5 Normal human karyotype (46XY). |
Metaphase chromosome preparations can be prepared from any cell undergoing mitosis. Typically, in order to obtain adequate numbers of cells, mitosis is induced artificially by using a mitogenic chemical such as phytohemagglutinin. The cells are then incubated in a dilute solution of an agent that poisons the mitotic spindle. The chromosomes are swollen by using a hypotonic salt solution, fixed on slide, and dried for staining. The stained chromosomes can be observed by light microscopy.
The dyes used to stain chromosome preparations reveal patterns of light and dark bands that reflect regional variations in the molecular composition of each chromosome. Giemsa is the most commonly used dye. Q-banding is a fluorescence technique that gives results similar to the G-banding (i.e., Giemsa banding), while R-banding (i.e., reverse banding) gives a pattern opposite to that with G- or
Q-banding. T-banding specifically stains the telemetric regions of chromosomes, which can help to screen for missing regions at the ends of the chromosomes, and C-banding primarily stains the centromeric region of the chromosomes.
Q-banding. T-banding specifically stains the telemetric regions of chromosomes, which can help to screen for missing regions at the ends of the chromosomes, and C-banding primarily stains the centromeric region of the chromosomes.
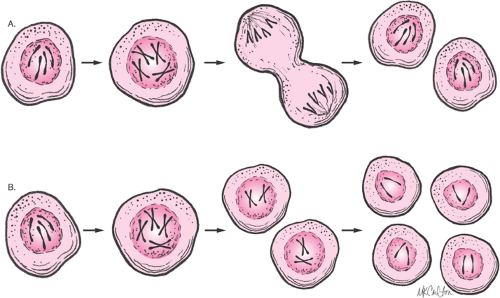 Figure 6.6 Mitosis and meiosis. The processes of mitosis (A) and meiosis (B) are complex and highly regulated. During mitosis, the chromosomes condense and attach to fibers that pull the sister chromatids to opposite sides of the cell. The cell then divides in cytokinesis, to produce two identical daughter cells. In meiosis, a second reduction division results in a haploid gamete—a gamete with only one member of each chromosomal pair. |
 Get Clinical Tree app for offline access 
|


