(1)
Department of Pediatric Dermatology, National Institute of Pediatrics, Mexico City, Mexico
Abstract
The major types of hair shaft defects are associated with genodermatoses and syndromes. Clinically, hair shaft defects may cause an unusual appearance, or a fragile short and sparse hair. Light microscopy of hair shaft defects are classified by their morphology and it is an important tool for diagnosis. Hair shaft disorders are separated by hair fragility into those with or without increased fragility. Those with increased fragility include monilethrix, pili torti, trichorrhexis nodosa, trichorrhexis invaginata and trichoschisis. Hair shafts without increased fragility include pili annulati, loose anagen hair, wooly hair and uncombable hair. Advances in the genetic causes of the hair shaft defects are described in the different entities, which have allowed to understand the mechanisms of the defects and to elucidate normal and pathogenic pathways. In patients with hair shaft abnormalities as an isolated defect, the main problem is aesthetical. In contrast, when the hair shaft defects are associated with syndromes the prognosis will depend on the associations of each condition, especially when metabolic, neurologic or cardiac defects are present.
Keywords
Hair shaft defectsMonilethrixPili tortiTrichorrhexis nodosaTrichoschisisTrichorrhexis invaginataPili annulatiLoose anagen hairUncombable hairBackground/Introduction
Hair follicles are ectodermally derived and produce hairs that range in size from minute vellus hair to long, thick terminal hair. The hair shaft is composed of three layers: the medulla, the cortex and the cuticle. Evaluation of the hair bulb phases, e.g. anagen, catagen and telogen, is relevant to determine the different causes of alopecia. Anagen hairs have a pigmented and indented elongated root. In the scalp, anagen follicles usually grow from 2 to 7 years, while shorter hairs and vellus hairs have shorter growth periods. Anagen follicles are actively replicating and, therefore, are more susceptible to nutritional and metabolic deficiencies. Anagen hairs are difficult to detach in a pull test and therefore when present one should suspect a hair shaft disorder. Catagen phase occurs with cessation of pigment formation and migration of the dermal papillae and follicular unit towards more superficial layers of the dermis. Catagen hairs represent 1 % of the scalp hairs. Telogen hairs have short, club-shaped roots and pigment is lacking. With the formation of new anagen hair below the club, the developing follicle will replace the telogen hair leading to shedding 50–100 scalp hairs a day. Telogen hairs represent 6–10 % of all terminal scalp hair and are easy to detach with a pull test without pain. A pull test is performed using a gentle traction on the patient’s hair; 4–6 or fewer hairs extracted is considered normal.
Epidemiology/Demographics
Genetic hair shaft defects may present in almost all races.
Epidemiology of the genetic hair shaft disorders varies according to the different syndromes.
A retrospective review performed on an Asian pediatric population was recently reported. Hair samples from 119 patients in a 10-year period were examined microscopically; half of the studied patients had abnormalities in the hair and one-third (31 %) showed morphologic changes compatible with specific diagnoses of various genetic conditions including 25 cases of loose anagen hair syndrome, six cases of uncombable hair syndrome, two cases of Netherton syndrome, three cases of Menkes syndrome and one case of trichothiodystrophy [1].
Clinical Presentation
The recognition of the anatomic characteristics of normal hair is important when evaluating a patient with hair abnormalities.
Light microscopy of the hair shaft and hair bulb is an important tool for the diagnosis of disorders affecting the hair.
Hair shaft abnormalities are divided into those with or without increased fragility.
Hair abnormalities can be seen in various genodermatoses and syndromes. One should suspect a hair shaft disorder if a patient presents with an abnormality or change in hair texture, color, appearance, manageability or ability to grow hair long. The initial evaluation includes a good history, physical examination and review of symptoms. A pull test determines whether there is hair breakage (increased fragility) by looking for broken hairs. An approach can be used to narrow the differential diagnosis in hair shaft disorders with and without fragility (Table 14.1). Light microscopy examination of the hair is necessary to evaluate the hair bulb and the microscopic structure of the hair shaft.
Table 14.1
Hair shaft disorders with increased fragility
Monilethrix |
• The term monilethrix refers to hair shafts with elliptical nodes at regular intervals. |
• The nodes have a diameter of normal hair and may be medullated, whereas the internodes are narrower and usually non-medullated, being the sites of fracture |
Clinically, patients with monilethrix usually present with diffuse alopecia with sparse, brittle short broken hairs on the entire scalp resulting from hair fragility over friction areas, predominantly the temporal and occipital regions. Other common findings are keratotic follicular papules at the nape of the head and keratosis pilaris, but a variable phenotypic expression and inheritance pattern are present. Hair rarely grows beyond 1–2 cm in length resulting in a stubbly appearance (Fig. 14.1a, b).
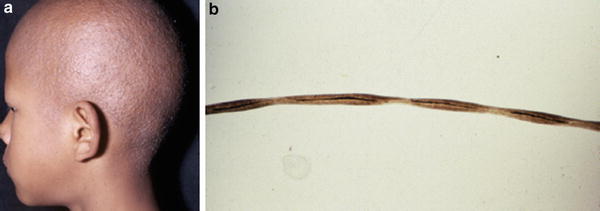
Fig. 14.1
(a) Monilethrix. Clinical appearance. (b). Hair shafts present elliptical nodes at regular intervals. ×20
Monilethrix is inherited as an autosomal dominant or autosomal recessive hair disorder with variable penetrance [2, 3]. Mutations are in the hair cortex keratins. The human keratin family comprises 54 members, 28 type I and 26 type II. Mutations causing autosomal dominant monilethrix have been found in the helix initiation and helix termination motifs of the type II hair keratins KRT81, KRT83, and KRT86 [4–6], while mutations in desmoglein 4 (DSG4) gene, which belongs to the desmosomal cadherin superfamily and is also expressed in the cortex of the hair follicle, are linked to recessive transmission [7].
Most monilethrix cases lack systemic involvement. Only isolated reports demonstrate association with congenital defects. A case report described atrophic alopecia associated with intractable scalp pruritus, diffuse keratosis pilaris, and bilateral posterior subcapsular cataracts and brachiocephaly, in a 9-year-old Turkish boy from consanguineous parents, suggesting an autosomal recessive trait [8]. Feng YG et al. reported a large Chinese pedigree with congenital monilethrix and hereditary unilateral external auditory canal atresia [9].
A diagnosis is elucidated by examining hairs by light microscopy observing the medullated nodes and non-medullated internodes in the hair shaft. Trichoscopy of scalp hair may reveal characteristic uniform elliptical nodes and intermittent constrictions along with variation in hair shaft diameter, presence of few vellus hairs and yellow dots. Dermoscopy of the keratotic follicular papules revealed multiple stubs of broken hair arising from them with a similar beaded appearance, suggesting a diagnosis of monilethrix [10].
There is no specific treatment that successfully cures the condition. A therapeutic trial with oral N-acetyl cysteine was attempted. There was slight improvement after 2 months of therapy. The hair density, however, did not show any further improvement subsequently [11]. Oral retinoids have been reported to obtain good clinical and cosmetic results while treatment was continued. In the past etretinate [12] and recently acitretin [13, 14] were reported to improve hair growth. However, clinical symptoms recurred after discontinuation and keratosis pilaris persisted. Topical 2 % minoxidil has been reported to increase the normal hair shaft without any side effects, after 1 year of treatment, and could be considered a good therapy [15].
Although there are no specific treatments prognosis is good.
Pili Torti
The term pili torti (PT) refers to a hair shaft which is flattened and twisted with an angle of 180°.
Fracture occurs within the twist, which is the weakest point.
Pili torti may occur as an inherited, isolated phenomenon or as a feature of different syndromes with neurological impairment, hearing loss or ectodermal dysplasias.
Pili torti may occur as an inherited, isolated phenomenon with onset in the early months of life. The hair is usually fairer than expected and is spangled, dry and brittle. Alterations of the inner root sheath likely lead to the abnormal molding and twisting of the hair shaft on its own axis. Inheritance patterns can be autosomal dominant [16], autosomal recessive [17] or sporadic [18]. A late onset of pili torti has been described with the onset in childhood or after puberty. It is an autosomal dominant condition in white patients with black unruly, coarse, stiff hair and non-progressive mental deficiency [19]. Other inherited entities may present pili torti and include Menkes syndrome, Björnstad syndrome, Crandall syndrome and ectodermal dysplasia.
Menkes kinky hair syndrome, also known as trichopoliodystrophy, is a rare X-linked recessive, progressive neurodegenerative with connective tissue manifestations. It is clinically characterized by progressive psychomotor impairment, treatment-refractory seizures and hair shaft abnormalities [20], most commonly PT, but other defects, such as trichorrhexis nodosa (TN), have been described. The condition is related to a mutation in a copper-transporting gene located on chromosome Xq13.3 resulting in a disorder caused by deficiency or dysfunction of a copper-transporting ATPase, ATP7A [21]. Four types of Menkes disease can be distinguished. The two extremes forms, the severe classical form and the mildest, called occipital horn syndrome (OHS), affect >90 % of patients. Moderate and mild forms likely represent variant of MD and OHS. The infants appear normal at birth and then typically develop neurologic deterioration, lethargy and a loss of milestones in the second or third months of age. Skin and hair hypopigmentation and bone and connective tissue alterations with soft skin and joint laxity are present. There is microcephaly and the hair shaft does not grow long and is easily broken. Light microscopy is an important clue for the early suspicion of Menkes syndrome. Flattened hair is twisted 180° along its axis intermingled with normal hair (Fig. 14.2). The diagnosis can be confirmed by a low plasma level of copper and ceruloplasmin. If untreated, most patients die within the first year of life. Longer-term survivors are reported as a result of early treatment with parenteral administration of copper in the neonatal period. It may prevent neurological deterioration but does not improve the non-neurological features such as hair or connective tissues laxity [22].

Fig. 14.2
Pili torti. Hair is twisted 180° along its axis intermingled with normal hair. ×20
Björnstad and Crandall Syndromes
Pili torti and hearing loss
In 1965, Björnstad described eight patients with pili torti and hearing loss [23]. The combination of these two findings was later coined Björnstad’s syndrome. Patients develop hair loss in the first 2 years of life, while the hearing deficit may become evident in the first 3–4 years of age. Only 39 cases of Björnstad syndrome have been reported in the English literature and only two cases have been reported in Japan [24].
Both autosomal and recessive inheritance patterns have been described and mapped to the gene locus 2q34–q36 [25]. Al-Owain et al. described a novel phenotype in a series of nine Saudi Arabian patients from four consanguineous families three of which were related. They identified a causative mutation in the BCS1L gene [26].
Crandall syndrome is similar to Björnstad syndrome but presents with hypogonadism [27]. Most cases are autosomal recessive.
Mental retardation is rarely associated in both syndromes.
Trichorrhexis Nodosa
The term TN refers to the light microscopic appearance of a fracture associated with swelling of cortical cells.
The hair is very fragile and it breaks readily with trauma or spontaneously.
TN may occur alone but has been reported in certain genodermatoses and metabolic disorders.
In congenital TN, inherited as an autosomal dominant trait, the hair is usually normal at birth but is replaced within a few months with abnormal, fragile hair. The condition tends to improve with age.
In two main metabolic inborn errors of the urea cycle, argininosuccinic aciduria and citrullinemia, TN can occur. TN occurs in approximately 50 % of cases of the neonatal form of argininosuccinic aciduria while in citrullinemia may present TN, atrophic hair bulbs, and/or pili torti. Clinically, manifestations are similar in both conditions; symptoms include lethargy, seizures, respiratory distress and development delay. Hair is usually normal at birth, with later development of dry, dull hair and TN. The light microscopic appearance of TN is a fracture associated with swelling of cortical cells and splaying them out from the main body of the hair shaft resembling the end of a brush (Fig. 14.3). The condition reverts to normal with dietary treatment of the metabolic condition, but it recurs quickly if the diet is abandoned [28].

Fig. 14.3
Trichorrhexis nodosa. Swelling of cortical cells causes a fracture resembling the end of a brush. ×40
Trichoschisis
The term refers to the light microscopic appearance of a sharp transverse fracture of the hair shaft (Fig. 14.4).

Fig. 14.4
Microscopic appearance of a sharp transverse fracture characteristic of trichoschisis. ×40
Under polarized light, the characteristic “tiger tail” pattern alternating bright and dark diagonal bands are observed.
Trichoschisis is a marker for a neuroectodermal disorder named trichothiodystrophy with brittle hair and low sulfur content of hair.
Trichothiodystrophy (TTD) is a heterogeneous rare group of autosomal recessive disorders of DNA repair unified by the presence of sulfur-deficient brittle hair. It is caused by a mutation of a regulatory gene involved in the transcription of DNA. There is a wide variety of phenotypes, from brittle hair only to severe intellectual impairment and developmental delay (Fig. 14.5).
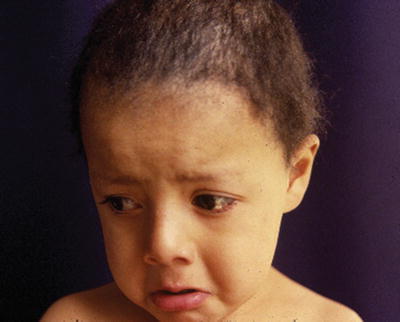
Fig. 14.5
Clinical aspect of a patient with trichothiodystrophy and mental retardation with brittle short hair
Eight subgroups have been categorized by Itin et al. [29] and include BIDS (brittle hair, intellectual impairment, decreased fertility, and short stature), IBIDS (BIDS + ichthyosis), PIBIDS (BIDS + photosensitivity), SIBIDS (otosclerosis + IBIDS), ONMR (onychotrichodysplasia, chronic neutropenia, and mental retardation), and Sabinas, Pollitt, and Marinesco–Sjögren syndromes [30]. Individuals with TTD may present with a collodion baby phenotype and intrauterine growth retardation can occur.
In TTD, 95 % of photosensitivity patients have abnormalities in nucleotide excision repair (NER) and can be assigned to the xeroderma pigmentosum (XP) complement group D (XPD) [29]. Non-photosensitivity TTD patients represent a genetically heterogeneous disorder. Mutations in chromosome 7p14 have been identified in two types of patients with Amish brittle-hair syndrome and non-photosensitivity TTD with mental retardation and/or decreased fertility [31].
The clinical appearance is a short, brittle, disordered sparse hair. This may be absent at birth and is not fully developed until 3 months of age [32]. On light microscopy, the hair has an irregular outline and a flattened shaft in which twists occur like a folded ribbon. Two types of fractures are seen—trichoschisis and an atypical trichorrhexis nodosa (less splaying out of the cortical cells). Under polarized light the typical “tiger tail” pattern confirms the diagnosis. The low cystine (sulfur) content of hair is postulated to account for cuticular and cortical weakness, provoking the fracture. A study reveals an inverse correlation between sulfur content and percent of hairs with shaft abnormalities (trichoschisis, trichorrhexis nodosa, or ribbon/twist), but there was no association between clinical disease severity and percent of abnormal hairs [33].
Diagnosis. Is made by visualization of the “tiger tail” banding seen in all hairs with polarizing microscopy providing a reliable diagnostic test. Scanning electron microscopy shows a flattened shaft with irregular ridging and disordered reduced, or absent cuticle scan pattern.
Trichorrhexis Invaginata
The term refers to a distal hair shaft invaginating into the proximal hair shaft (“bamboo hair”).
Trichorrhexis invaginata (TI) is the characteristic hair abnormality of Netherton syndrome.
Netherton syndrome (NS) is a severe autosomal recessive skin disorder characterized by congenital ichthyosiform erythroderma, TI, and atopic manifestations with an elevated IgE level [34]. Recently, pathogenic mutations were identified in serine protease inhibitor Kazal-type 5 (SPINK5) [35], the gene that encodes lympho-epithelial Kazal-type related inhibitor (LEKTI), a type of serine protease inhibitor involved in the regulation of skin barrier formation and immunity. The major neonatal complication is the hypernatremic dehydration and staph skin infections complicated by neurologic signs.
Although the severity varies considerably, the clinical and microscopic findings are present from birth with a large variability in phenotypic expression. Most patients present a congenital generalized exfoliative erythroderma which can last up to 2 years followed by an ichthyosiform eruption with polycyclic erythematous plaques with fine double-edged scaling, named ichthyosis linearis circumflexa (Fig. 14.6).
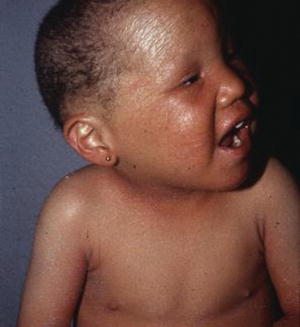
Fig. 14.6




Netherton syndrome in a patient with congenital ichthyosiform erythroderma. Hair is sparse as well as eyebrows and eyelashes. Courtesy Prof. R. Ruiz-Maldonado
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
