Fig. 2.1
A laparoscopic image of a uterus in reproductive age with a fundal myoma
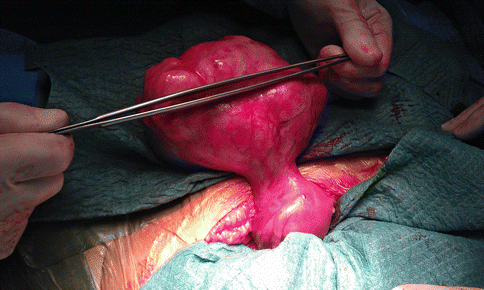
Fig. 2.2
A laparotomic image of a large fundal pedunculated myoma, as a benign smooth muscle uterine tumor that occur via clonal expansion from a single mutated myometrial smooth muscle stem cell
In the recent years, significant progress has been made in our understanding of myomas tumorigenesis. A current model suggests that a distinct stem/reservoir cell-enriched population, designated as the leiomyoma-derived side population (LMSP), is responsible to sustain proliferation and tumor growth [3].
Myomas are classified by their location relative to the layers of the uterus (as subserous, intramural, or submucous) and can occur as single or multiple tumors of varying size (Fig. 2.3) [4].
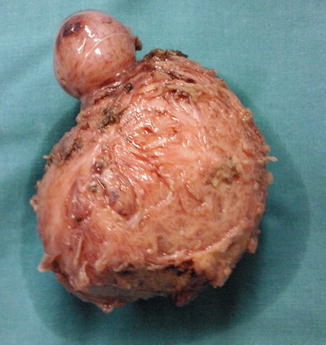
Fig. 2.3
An enucleated myoma composed by two tumors, one smallest and one larger
Leiomyomas are often asymptomatic but can cause a multitude of symptoms such as abnormal uterine bleeding, a feeling of pelvic pressure, urinary incontinence or retention, or pain [5].
The exact pathophysiology of uterine leiomyomas is still unknown, however several epidemiologic studies have linked uterine leiomyomas to different risk factors including high levels of female hormones (estrogens and progesterone), family history, African ancestry, early age of menarche and obesity. In contrast, it was found that childbearing at a later age is inversely associated with the risk of developing leyomyomas. There has been recent evidence suggesting a relationship between alcohol and caffeine intake with a risk of developing fibroids. Metabolic, dietary, stress, and environmental factors may also play a role in fibroid development [5].
The traditional surgical options for myomas are hysterectomy and myomectomy, nonsurgical medical therapies are available but often ineffective in eliminating myomas and preventing recurrence.
Hormones have been considered as the major promoter of leiomyoma growth. In addition, several pathogenetic factors such as genetics, microRNA, growth factors, cytokines, chemokines have a role in the development and growth of the disease [6].
The mechanical properties of leiomyoma are another key feature of these tumors that may contribute to their growth. Leiomyomas are in fact stiff tumors characterized by an excessive deposition of disordered extracellular matrix (ECM) components, particularly collagen I, III, and IV, proteoglycans, and fibronectin [6]. Matrix metalloproteinases (MMPs) are implicated in leiomyoma remodelling with a higher activity of MMP-2 in leiomyomas than in surrounding myometrium [7]. Myomas are also surrounded by a thin fibro-neurovascular pseudocapsule, which separates myomas from normal peripheral myometrium [8]. This means that the tumor microenvironment may greatly influence tumor growth and proliferation.
Molecular Aspect of Uterine Leiomyomas
Uterine fibroid is a multifactorial and still enigmatic pathology. Classic studies showed steroid dependence of myomas for growth and development. The genetic background seems to play an important role, with cytogenetic anomalies observed in about 40 % of uterine fibroids. Abnormal ECM expression, increased growth factors, cytokines and chemokines concentrations, and an extracellular disorganized matrix have been implicated in development and growth of uterine leiomyomas [9].
Most molecular studies of myomas have focused on the analysis of few genes or taken into account alterations in specific signalling pathways. These studies have revealed alterations of genes/proteins related to proliferation, cell-cycle regulation, apoptosis, and cell-cell adhesion.
Estrogens may exert their growth-stimulatory effects on leiomyomas through the action of a complex network of cytokines, growth factors, or apoptosis factors and through different cellular mechanisms [10]. In experiments with animal models, Ishikawa and collaborators suggested that estrogens induce the expression of the progesterone receptor (PR), thus supporting the action of progesterone on leiomyoma xenografts. Furthermore, estrogens may stimulate leiomyoma growth partially by suppressing normal p53 functions [11]. Biochemical and clinical studies also suggested that progesterone, progestins, and progesterone receptors (PR-A and PR-B) might increase proliferative activity in leiomyomas by enhancing the expression of growth factors (EGF, IGF-I) and apoptosis-related factors (TNFalpha, Bcl-2 proteins) [12].
Uterine myomas cells typically show a high expression of cell-cycle regulator and anti-apoptotic proteins. This can trigger tumor growth and make cells resistant to apoptosis.
Lora and collaborators demonstrated that the ratio between PR-A and PR-B is similar in normal myometrium and leiomyomas while p53 and p21 mRNA and protein levels are increased in leiomyomas [13]. Matsuo and collaborators showed that Bcl-2 protein, an apoptosis- inhibiting gene product, was abundantly expressed in myomas compared with normal myometrium. In this study, Bcl-2 protein expression in myoma cells was up-regulated by progesterone, but down-regulated by estradiol. The same group reported up-regulation of expression of proliferating cell nuclear antigen (PCNA) in myomas by progesterone and estradiol [14].
Wang and collaborators showed that protein and mRNA expression of bFGF and T-cadherin in uterine leiomyoma were present with significantly higher expression than that in adjacent normal myometrium and control normal myometrium. In addition, T-cadherin correlated well with bFGF. There was a relationship between T-cadherin and color Doppler flow imaging (CDFI) [15].
Epidemiological differences observed between different ethnic groups have a strong association with the genetic background. This is illustrated by a large-scale gene and protein expression-profiling study that provided valuable information about the molecular alterations of leiomyoma and myometrium in Caucasians and African Americans [16]. Data from genomic and proteomic studies demonstrated that many of the differences in leiomyoma’s gene expression observed in the two ethnic groups might be attributed to differences in myometrial gene expression, as well as differences in leiomyomas vs. myometrium. Moreover, functional analysis of microarray and proteomic data revealed that many of the observed differences may be attributed to molecules with a role as transcriptional, translational and signal transduction mediators, cell cycle and EMC regulators, cell-cell adhesion and metabolic regulators. The current approach to diagnosis and treatment should evolve in the future and consider women with a greater genomic risk.
The Genomic Landscape of Uterine Myomas: Mutation Analysis and Chromosome Rearrangements
Several clinical evidences that allow the molecular characterization of this tumor support the presence of genetic mechanism involved in fibroids aetiology.
Somatic mutations involving the gene encoding the mediator complex subunit 12 (MED12) and the gene encoding the high-mobility group AT-hook 2 (HMGA2) are known to be associated to leiomyoma [1].
Mäkinen and collaborators [17] found that approximately 70 % of tumors contained heterozygous somatic mutations that affect MED12, a gene locates on the X chromosome. Authors described that all mutations resided in exon 2 (codon 44), suggesting that aberrant function of this region of MED12 contributes to tumorigenesis. Moreover, they also performed a pathway analysis, comparing eight tumors positive for MED12 mutations with their respective normal tissues. Three pathways were found to be substantially altered in the tumors namely, focal adhesion, extracellular matrix receptor interaction, and Wnt signaling pathways. This suggests that MED12 mutations contribute to tumor development by altering specific cellular pathways. The association between MED12 and the Wnt pathway is also supported by the study of Markowski and collaborators which showed a significant upregulation of a member of Wnt pathway, Wnt4, in fibroids with MED12 mutation compared to those with HMGA2. Wnt4 is known to be expressed in the mesenchyme of the Müllerian duct giving rise to the likely tissue of origin of uterine leiomyomas. The overexpression of Wnt4 in the group of fibroids with mutations of MED12 compared to tumors with HMGA2 rearrangement suggests that Wnt4 may be considered as a possibly relevant downstream effector of the mutated MED12 [18].
MED12 belongs to a family of evolutionarily conserved transcriptional factors (Mediator) that promote the assembly, activation, and regeneration of transcription complexes on core promoters during the initiation and reinitiation phases of transcription (Fig. 2.4).
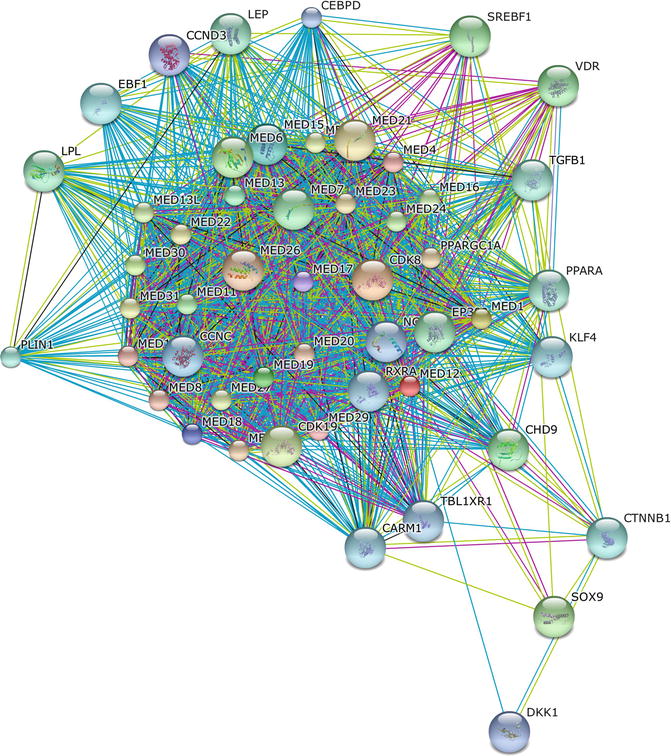
Fig. 2.4
The protein-protein interaction network of MED12 was determined using the online software STRING. The network nodes are proteins. The edges represent the predicted functional associations. An edge may be drawn with up to seven differently colored lines – these lines represent the existence of the seven types of evidence used in predicting the associations. A red line indicates the presence of fusion evidence; a green line – neighborhood evidence; a blue line – co-ocurrence evidence; a purple line – experimental evidence; a yellow line – textmining evidence; a light blue line – database evidence; a black line – co-expression evidence. Predicted functional partners include several members of the MED protein family, as well as transcriptional factors, microRNA, growth factors, and metabolic regulators
In detail, this gene codifies for a 26-subunit transcriptional regulator that bridges DNA regulatory sequences to the RNA polymerase II initiation complex. It is a subunit of the “kinase” module of the mediator complex, which also contains MED13, CYCLIN C, and Cyclin-dependent kinase 8 (CDK8). MED12 is frequently mutated in human cancers including prostate [19], and renal cell carcinomas [20].
It was also observed that activation of ERK signaling by MED12 suppression, may confer resistance to tirosin kinase inhibitors including crizotinib, gefitinib, vemurafenib, seluteminib, and sorafenib, thus providing a link between suppression of MED12 and drug resistance. Data also indicate that MED12 suppression induces an epithelial mesenchymal transition (EMT)-like phenotype and that this EMT-like phenotype induced by MED12 KD is associated with chemotherapy resistance in both cell lines and patients [21].
Consistent with their role in the regulation of gene transcription, members of the Mediator family are functionally required for activated transcription in response to diverse cell signaling pathways. These include MED1 (TRAP220, ARC/DRIP205) for nuclear receptor, MED14 (TRAP170, ARC/DRIP/CRSP150 (cofactor required for Sp1 function) for interferon-γ, MED23 (TRAP150β, ARC/DRIP/CRSP130, human SUR2) for Ras/mitogen-activated protein kinase (MAPK), and MED15 (ARC105, PCQAP) for Transforming growth factor-β (TGF-β) signaling pathways [22, 23]. MED12 binds directly to β-catenin and regulates canonical WNT signaling [24]. Mechanism involving MED12 mutations, WNT–β-catenin activation, and hyperactive TGF-β signaling supports stem-cell renewal, cell proliferation, and fibrosis in uterine fibroid tissue [25–27]. MED12 has also been shown to modulate the sonic hedgehog (Shh) signaling pathway through Gli3 interaction [28]. As demonstrated by Mäkinen and collaborators, MED12 mutations alone are sufficient for driving tumor development. Authors analysed whole exome sequencing data of 27 uterine leiomyomas (12 MED12 mutation-negative and 15 MED12 mutation-positive) and their paired normal myometrium. They searched for genes, which would be recurrently mutated. No such genes were identified in MED12 mutation-negative uterine leiomyomas as well as MED12 mutation-positive leiomyomas. These results highlight the unique role of MED12 mutations in genesis of uterine leiomyomas [29]. Moreover, it was recently demonstrated that pelvic and retroperitoneal leiomyomas harbor an increased frequency of MED12 mutations (34 %) as compared with other extrauterine sites (0 %; P = 0.0006), and that histologically unremarkable adjacent myometrium can harbor similar MED12 mutations suggesting that smooth muscle tumors in pelvic/retroperitoneal sites are subject to the same mutational changes as those of uterine myometrium [30].
Although leiomyomas are belived to be chromosomally rather stable, cytogenetic rearrangements have been detected in 40–50 % of leiomyomas. Studies found translocation between chromosomes 12 and 14, trisomy 12, translocation between chromosomes 6 and 10 and deletion of chromosomes 3 and 7, with multiple candidate genes [31–34]. Deletion of 7q, rearrangements involving 12q15 and 6p21 occur in 17, 20 and 5 % respectively [35, 36]. HMGA2 is the driver gene for tumor carrying 12q15 rearrangements while high-mobility group AT-hook 1 (HMGA1) is involved in 6p21 rearrangements. The targeted translocation partner of HMGA2 (and HMGA1 in some cases) in leiomyoma is chromosomal band 14q24 [37–39]. The 14q24 breakpoint maps to a recombinational repair gene (RAD51B) locus [40], and the most frequent anomaly is t(12;14)(q14-15;q23-24).
HMGA2 is a protein that belongs to the non-histone chromosomal high mobility group (HMG) protein family which are often referred to as architectural proteins, participate in a wide variety of cellular processes including regulation of inducible gene transcription, integration of retroviruses into chromosomes, and the induction of neoplastic transformation and promotion of metastatic progression of cancer cells (Fig. 2.5).
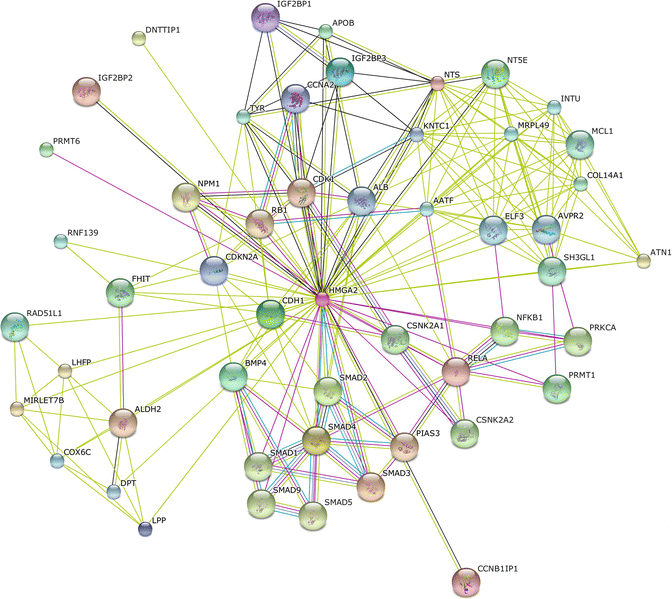
Fig. 2.5
The protein-protein interaction network of HMGA2 was determined using the online software STRING. The network nodes are proteins. The edges represent the predicted functional associations. An edge may be drawn with up to seven differently colored lines – these lines represent the existence of the seven types of evidence used in predicting the associations as described before. Predicted functional partners include several members of the SMAD protein family, PKC, microRNAs, cell cycle and metabolic regulators
They are characterized by the presence of 3 copies of a conserved DNA-binding peptide motif (AT-hook) that preferentially binds with the minor groove of many AT-rich promoter and enhancer DNA regulatory elements. Protein-protein and protein-DNA interactions induce both structural changes in chromatin substrates and the formation of stereospecific complexes called ‘enhanceosomes’ on the promoter/enhancer regions of genes whose transcription they regulate [41, 42].
Aberrations in the chromosomal region 12q14-15 that affect HMGA2 are frequent in a variety of tumours. In benign tumours of mesenchymal origin, HMGA2 is often rearranged by translocation, and the resulting chimeric transcripts are formed by fusion of the DNA-binding domains, coded by exons 1–3, to ectopic sequences [43–45]; thus losing the C terminus and the 3′UTR region. In sarcomas, HMGA2 is frequently and selectively amplified and rearranged [46].
The HMGA2 3′-UTR contains target sites for the let-7 miRNA, and thus the above mentioned rearrangements lead to over-expression of HMGA2 protein due to loss of miRNA-mediated repression [47, 48]. Furthermore, it appears that the balance between let-7 and HMGA2 governs the exit of cells from the undifferentiated and self-renewing state, and HMGA2 is now thought to be central in cancer in general [49–52]. HMGA2 overexpression may favor self-renewal of mutated cells and offset senescence by suppressing cyclin-dependent kinase inhibitor 2a (Cdkn2a), which encodes the proteins p16Ink4a and p14Arf, negative regulators of their self-renewal [53]. Intriguingly, uterine fibroids are deficient in the let-7 miRNA that targets and suppresses HMGA2 [54]. Kumar and collaborators [55] recently demonstrated that the oncogenic potential of the HMGA2 gene is largely due to the ability of its transcript to operate as a competing endogenous RNA (ceRNA) in a protein coding-independent manner. The HMGA2 mRNA decoys the let-7 microRNA family to regulate Tgfbr3 expression and enhance TGF-β signaling, thereby promoting lung cancer progression.
Most recently, the development of high informative genomic platforms provides further opportunities to characterise the molecular portrait of this tumor.
Mehine and collaborators performed a characterization of uterine leiomyoma by whole genome sequencing and gene expression profiling which showed that interconnected complex chromosomal rearrangements (CCRs) resembling chromothripsis (a single genomic event that results in focal losses and rearrangements in multiple genomic regions) were a common feature of leiomyoma and occurred in the presence of normal TP53 alleles [56]. These rearrangements are best explained by a single event of multiple chromosomal breaks and random reassembly, which are a major cause of chromosomal abnormalities in uterine leiomyoma. The rearrangements appear responsible of tissue-specific changes consistent with a role in the initiation of leiomyoma. Rearrangements between chromosome 12 and 14, combining the 5′ end RAD51B with the full length high-mobility group AT-hook 2 (HMGA2), have been detected as a CCR event, and provide evidences that translocated RAD51B acts as an effective enhancer HMGA2. Further CCRs have been identified as translocation of HMGA2 to chromosome 5, or as breakpoint in chromosome 12 that removes the HMGA2 target sequence for the microRNA repressor let-7b, providing a mechanism of up-regulation of HMGA2. Other CCRs involving chromosomes 5 and 6 rearranged the full length HMGA1 to the MIR143HG, encoding a precursor of the microRNAs miR-143 and miR-145, which regulate the differentiation of smooth-muscle cells [57]. Mehine and collaborators also identified aberration affecting collagen genes COL4A5 and COL4A6 on chromosome X22q. This locus is also constitutively disrupted in person with Alport’s syndrome and diffuse leiomyomatosis [58] and it was associated of higher expression of insulin-receptor substrate 4 (IRS4) in leiomyoma [56]. In the same study CCRs resulting in deletion of 7q, which are common in uterine leiomyoma, have been associated to several rearrangements at various sites for target genes involved in response to DNA damage (CUX1) or CDK6-driven cell-cycle arrest at the G1 phase (ZNHIT1) or in the ubiquitine-dependent degradation of cell-cycle regulator (CUL1). Furthermore in the same study they found that MED-12 mutated tumors had relatively few chromosomal aberration while trancriptome analysis showed an up-regulation of RAD51B which further suggests a role of the gene in the pathogenesis of leiomyoma [56].
In different studies, other genes have been found to be implicated in leiomyoma development. Cha and colleagues [59] genotyped 1,607 individuals with uterine fibroids and identified 3 susceptibility loci associated with uterine fibroids. Chromosome 10q24.33 seems to have the best association with leiomyomas; the region was mapped to the 5′ region of the SLK gene encoding STE20-like kinase. STE20-like kinase has a role in myogenic differentiation, and, after activation by epithelial disruption, it is expressed in proliferating myoblasts. Another gene product located in the region is A-kinase anchor protein-13 (AKAP13), associated with cytoskeletal filaments. Related mutations could alter the regulation of extracellular matrix deposition and, consequently, of the fibrotic phenotype of the leiomyoma [60].
In addition to somatic mutations, heritable cancer syndromes can be characterized by uterine leiomyomas such as hereditary leiomyomatosis and renal cell cancer (HLRCC). This syndrome predisposes patients to benign leiomyomas of skin and uterus and early-onset renal cell carcinoma. The syndrome is caused by heterozygous germline mutations in the gene encoding fumarate hydratase (FH), an enzyme of the tricarboxylic acid cycle. Somatic FH mutations have been also reported in a small subset (1.3 %) of sporadic leiomyomas. Both hereditary and sporadic tumors are characterized by biallelic loss of FH, which would be predicted to cause severe metabolic stress [61, 62].
Epigenetics in Myomas
Epigenetic mechanisms are essential for normal development as well as for cancer initiation and progression. DNA methylation is one of the most commonly epigenetic events taking place in the mammalian genome and aberrant DNA methylation is a prominent characteristic of human cancer. This process affects gene activity by a covalent chemical modification, resulting in the addition of a methyl (CH3) group at the carbon 5 position of the cytosine ring. CpG dinucleotides are concentrated across the human genome in short CpG-rich DNA stretches called CpG islands.
DNA methylation is catalysed either by maintenance methyltransferases (DNMT1) or by de novo methyltransferases (DNMT3a and DNMT3b) that are active after completation of replication [63]. A global hypomethylation and differential expression of DNMT1, 3a and 3b, were revealed in uterine leiomyoma as compared with the matched normal myometria raising the possibility that epigenetic mechanisms such as DNA methylation and histone modification may contribute to the development of these tumors [64].
Several studies reported an aberrant promoter methylation of myomas-related genes such as ER-α (80 %), DAP kinase (54 %), RASSF1A (39 %), p16INK4a (22–25 %), and MLH1 (6 %) [65–67] suggesting that global hypomethylation mechanisms may contribute to elevating the expression of these genes. Gloudemans and collaborators observed an inverse correlation between CpG methylation and expression of the insulin-like growth factor II (IGF-II) gene in malignant smooth muscle tissues. In normal smooth muscle and in leiomyomas the IGF-II gene appeared to be methylated, while in leiomyosarcomas with high IGF-II gene expression, the overall methylation of IGF-II gene tended to be low or absent [68].
Genomewide profiling of DNA methylation and messenger RNA (mRNA) expression in uterine fibroid tissue and matched normal myometrial tissue displayed hypermethylation at promoter sites that were associated with their silencing in the fibroid tissues [69]. In detail, authors identified a total of 585 transcriptional regulatory regions that were hypermethylated, and 446 were hypomethylated in uterine leiomyoma compared with adjacent normal myometrial tissue. They also considered the mRNA expression of these genes and observed that a total of 55 genes showed both differential DNA methylation and changes in mRNA expression in uterine leiomyoma and adjacent normal myometrial tissue.
MicroRNAs (miRNAs) are a class of class of 18–24 nucleotide RNA molecules that regulate a high number of biological processes by targeting mRNAs for cleavage or translation repression. A direct link between miRNA expression and cancer pathogenesis is supported by several studies examining the expression of miRNAs in tumor samples [70].
The regulatory role of these small RNA molecules has recently begun to be explored in myomas, where several miRNAs such as let7, miR-21, miR-34a, miR125b, miR-93, miR-106b, and miR-200 are significantly dysregulated in uterine leiomyoma compared to those in normal myometrium [71, 72]. Global analysis of miRNA expression in leiomyomas from different racial groups showed that 31 miRNAs were expressed differently among different races. In particular, miR-21, miR-23a/b, miR-27a, miR-197, miR-203, miR-411, and miR-412 are significantly overexpressed in the leiomyomas of African-American women when compared with that in Caucasian women [71]. This suggests that a miRNA profile or, in general, a different molecular signature as described above, may account for the high morbidity rate of myomas in African-American women compared with women from other races.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


