Chapter 43
Endocrine Disorders
Richard N. Wissler MD, PhD
Chapter Outline
Diabetes Mellitus
Definition and Epidemiology
Diabetes mellitus (DM) is a common metabolic disorder with a prevalence of 6.8% to 8.2% in the general adult population in the United States.1,2 DM results from either an absolute deficiency in insulin secretion (type 1) or a combination of resistance to insulin in target tissues and inadequate insulin secretion (type 2).3 Although a combination of genetic and environmental factors contributes to both types, type 1 DM is primarily an autoimmune disorder. Type 2 DM occurs primarily in obese individuals and accounts for 90% to 95% of cases of DM in the United States.3 Gestational DM refers to DM or glucose intolerance that is first diagnosed during pregnancy. Gestational DM occurs in approximately 7% of pregnancies in the United States, reflecting a doubling of its prevalence between 1990 and 2000.4,5
Pathophysiology
Insulin is a peptide hormone secreted by the beta cells of the islets of Langerhans in the pancreas. Insulin binds to specific cell-surface receptors in insulin-responsive target tissues (e.g., liver, skeletal muscle, fat). The intracellular effects of insulin are mediated by tyrosine kinase in the beta-subunit of the receptor through a cascade of distal protein kinase–mediated phosphorylations.6,7 Normal hepatic glucose metabolism represents a balance between the effects of insulin and several “counterregulatory” hormones (e.g., glucagon, cortisol, epinephrine, growth hormone).8 This control system for glucose homeostasis permits rapid adjustments in glucose metabolism in the fed and fasted states. Insulin is also an important anabolic regulator of lipid and amino acid metabolism (Figure 43-1). Insulin deficiency (absolute or relative) associated with DM results in abnormal metabolism of carbohydrates, lipids, and amino acids.
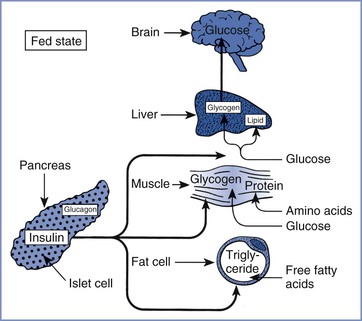
FIGURE 43-1 Substrate use in the fed state, showing the role of insulin in the promotion of fuel storage. (From Kitabchi AE, Murphy MB. Diabetic ketoacidosis and hyperosmolar hyperglycemic nonketotic coma. Med Clin North Am 1988; 72:1545-63.)
Acute and chronic complications occur in patients with DM (Box 43-1). The three major acute complications are diabetic ketoacidosis, hyperglycemic nonketotic state, and hypoglycemia. Diabetic ketoacidosis (DKA) occurs predominantly in patients with type 1 DM. DKA may develop with a new source of insulin resistance (e.g., infection, trauma, stress) and/or as a result of failure to administer usual insulin doses. DKA results from decreased uptake of glucose by insulin-responsive tissues and greater use of free fatty acids as a hepatic energy source. The lack of insulin favors lipolysis, beta-oxidation of free fatty acids in the liver, and hepatic formation of acetoacetate and beta-hydroxybutyrate from the excess acetyl-coenzyme A generated by fatty acid oxidation.9 These biochemical events result in metabolic acidosis, hyperglycemia, and dehydration secondary to osmotic diuresis. Signs and symptoms of DKA include nausea, vomiting, weakness, tachypnea, hypotension, tachycardia, stupor, and acetone on the breath. The diagnosis of DKA depends on the laboratory findings of hyperglycemia, ketosis, and acidosis.10
Hyperglycemic nonketotic state (HNS) occurs predominantly in patients with type 2 DM. Laboratory findings in HNS are hyperglycemia (blood glucose level often > 600 mg/dL), hyperosmolarity (> 320 mOsm/kg), and moderate azotemia (serum blood urea nitrogen [BUN] often > 60 mg/dL), without ketonemia or significant acidosis.10 The absence of significant ketosis in HNS may indicate an inhibition of lipolysis by hyperosmolarity or low levels of insulin. DKA and HNS are probably related conditions; inadequate insulin therapy and infection are the most common precipitating events for both.10
Hypoglycemia is a continuing health threat in diabetic patients, especially in patients receiving insulin therapy. Hypoglycemia results from an imbalance between insulin or oral hypoglycemic agents and available metabolic fuels. In hospitalized patients with DM, major risk factors for hypoglycemia include renal insufficiency and decreased caloric intake.11 Symptomatic awareness of hypoglycemia and counterregulatory responses may be inadequate in some diabetic patients with autonomic neuropathy.12 Problems with hypoglycemia awareness in patients receiving beta-adrenergic receptor antagonists can be minimized by using β1-adrenergic receptor–selective antagonists.13 Factitious hypoglycemia results from a deliberate, inappropriate self-administration of insulin or an oral hypoglycemic agent.14
In general, the prevalence of chronic complications increases with the duration of DM.4,15 The Diabetes Control and Complications Trial, a randomized multicenter study of patients with type 1 DM, demonstrated a positive relationship between tight glucose control and a lower incidence or rate of progression of retinopathy, nephropathy, and neuropathy.16 In a similar study of patients with type 2 DM—the U.K. Prospective Diabetes Study (UKPDS)—intensive glucose control lowered the incidence of microvascular complications but not of macrovascular complications or patient mortality.17 In contrast, antihypertensive therapy reduced the incidence of macrovascular complications and mortality in patients with both type 2 DM and chronic hypertension.17 DM may affect cardiovascular function as a result of coronary atherosclerosis, autonomic neuropathy, or development of a cardiomyopathy.18
Clinical Presentation and Diagnosis
Box 43-2 lists the current diagnostic criteria for DM in nonpregnant patients.4
Gestational DM is associated with (1) advanced maternal age, (2) obesity, (3) family history of type 2 DM, (4) prior history of gestational DM, (5) history of polycystic ovarian syndrome, (6) glycosuria, and/or (7) history of prior stillbirth, neonatal death, fetal malformation, or macrosomia. The clinical sensitivity of the medical history in detecting gestational DM is only 50%.19 Box 43-3 lists the current recommendations of the American Diabetes Association (ADA) for screening and diagnosis of gestational DM.4
Several observational clinical studies, including the Hyperglycemia and Adverse Pregnancy Outcome Study,20 have shown that adverse pregnancy outcomes are a continuous function of glucose intolerance in pregnancy. Based on these observations, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) has proposed new diagnostic criteria for gestational DM as described in Box 43-3.21 In this system, gestational DM is diagnosed if even one of the three blood glucose samples is elevated in a 2-hour oral glucose tolerance test. The ADA has approved the more inclusive IADPSG diagnostic criteria4; the new criteria are estimated to increase the diagnosed incidence of gestational diabetes to 16% to 18% of pregnant women.22 However, the American College of Obstetricians and Gynecologists (ACOG) has not accepted the new criteria of the IADPSG/ADA. The ACOG continues to recommend a two-step diagnostic process; screening all women at 24 to 28 weeks’ gestation followed by a 100-g, 3-hour oral glucose tolerance test for those who screen positive. With this conservative approach, the incidence of gestational DM is approximately 7%.23 The current controversy between the IADPSG/ADA and the ACOG diagnostic criteria for gestational diabetes is centered on whether treatment of an expanded patient population is cost-effective and will improve outcomes.22–26
Glycosylated hemoglobin measurements are used as time-integrated estimates of glycemic control but not as a diagnostic test for DM. The normal range for hemoglobin A1c in nondiabetic pregnant women is 4.0% to 5.5%, compared with 4.8% to 6.5% in nondiabetic nonpregnant women.27
Interaction with Pregnancy
How Does Pregnancy Affect Diabetes Mellitus?
Pregnancy is characterized by progressive peripheral resistance to insulin at the receptor and postreceptor levels in the second and third trimesters (Figure 43-2).28-30 The presumed mechanism involves an increase in counterregulatory hormones (e.g., placental lactogen, placental growth hormone, cortisol, progesterone) during pregnancy. The change in placental lactogen is a plausible mechanism, given that (1) a graph of serum lactogen levels during pregnancy is similar in shape to that of insulin requirements in pregnant women with type 1 DM and (2) placental lactogen has growth hormone–like activity. Also, maternal adipokines probably are important factors in insulin resistance of pregnancy29; they facilitate the provision of maternal fuels for the fetus.30
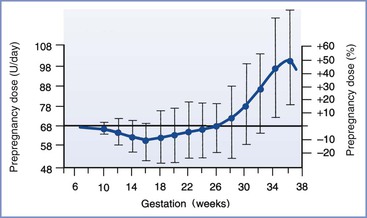
FIGURE 43-2 Insulin requirements in euglycemic women with type 1 diabetes mellitus during pregnancy. (From Crombach G, Siebolds M, Mies R. Insulin use in pregnancy: clinical pharmacokinetic considerations. Clin Pharmacokinet 1993; 24:89-100.)
Gestational DM develops when a patient cannot mount a sufficient compensatory insulin response during pregnancy. In some patients, gestational DM can be viewed as a preclinical state of glucose intolerance that is not detectable before pregnancy. After delivery most patients return to normal glucose tolerance but remain at increased risk for DM (predominantly type 2) in later life.31 The recurrence rate for gestational DM in a subsequent pregnancy is 35% to 70%.32
In patients with pregestational DM, insulin requirements progressively increase during pregnancy because of peripheral insulin resistance.33 At term, the daily insulin requirement is approximately 1.0 insulin unit/kg, compared with 0.7 unit/kg before pregnancy.33 Insulin requirements may be higher in pregnancies with multiple gestation.34 During late pregnancy in normal healthy patients, basal and glucose-stimulated plasma insulin levels are twice the postpartum measurements.30 These changes reflect pregnancy-related increases in pancreatic islet cell mass and glucose sensitivity, probably secondary to the net effect of competing progesterone and lactogenic hormone stimuli in the endocrine pancreas.35,36 Near term, maternal overnight insulin requirements may decrease, presumably as a result of a “siphoning of maternal fuels” by the growing fetus during the overnight maternal fast.37
Endogenous plasma insulin concentrations during labor and delivery in nondiabetic parturients differ from exogenous insulin requirements in laboring diabetic women. In nondiabetic parturients, the plasma glucose concentration is only one of many factors that affect endogenous insulin secretion; glucose production and use are markedly higher during painful labor than postpartum.38 Plasma insulin concentrations remain unchanged except for a brief increase during the third stage of labor and immediately postpartum.38,39 This finding suggests that glucose use during labor is largely independent of insulin. The patterns of plasma insulin concentrations are similar in nondiabetic patients with and without analgesia (e.g., nitrous oxide, meperidine).39
In patients with type 1 DM, insulin requirements decrease with the onset of the first stage of labor.40 These patients may require no additional insulin during the first stage of labor, although insulin requirements are modified by (1) the level of metabolic control before labor, (2) the residual effect of prior doses of subcutaneous insulin, and (3) the glucose infusion rate.40,41 Insulin requirements increase during the second stage of labor via an unknown mechanism.40,41 The use of epidural analgesia or oxytocin does not affect exogenous insulin requirements during the first and second stages of labor.40 After delivery—either vaginal or cesarean—insulin requirements in women with type 1 DM decrease markedly for at least several days, although there is significant variability among individuals (Figure 43-3).28,42 Presumably, the decreased insulin requirement results from loss of counterregulatory hormones produced by the placenta. Pituitary growth hormone responsiveness to hypoglycemia is blunted in late pregnancy and may contribute to impaired counterregulatory responses during the postpartum period.43 Insulin requirements gradually return to prepregnancy levels within several weeks of delivery in women with type 1 DM.37
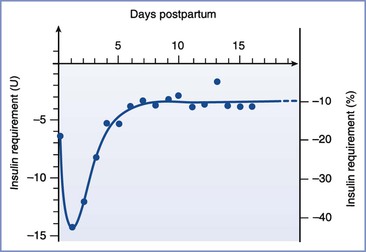
FIGURE 43-3 Insulin requirements in the postpartum period. (From Crombach G, Siebolds M, Mies R. Insulin use in pregnancy: clinical pharmacokinetic considerations. Clin Pharmacokinet 1993; 24:89-100.)
Before the discovery of insulin in 1921, pregnancies were rare in diabetic patients. Insulin therapy improved the rate of survival in women with severe DM, allowing these women to reach childbearing age and become pregnant. Maternal outcomes improved, but fetal and neonatal morbidity and mortality remained high.44
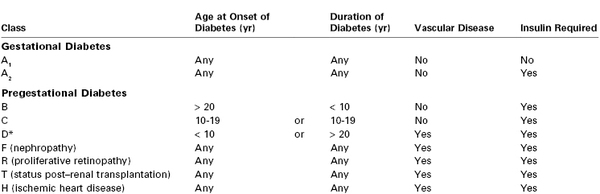
In 1949, White45 proposed a classification system for DM during pregnancy based on 439 consecutive cases. Physicians caring for pregnant diabetic patients should be familiar with the White system, which has endured with some modifications (Table 43-1). The system emphasizes the relationship among the duration of type 1 DM, vascular complications of type 1 DM, and poor fetal outcome.46 In the 1950s, fetal survival rates were as follows: class A, 100%; class B, 67%; class C, 48%; class D, 32%, and class F, 3%.46
Diabetic Ketoacidosis.
The incidence of DKA has decreased from 9% to between 1% and 2% of diabetic pregnancies,47,48 probably as a result of improvements in medical care and patient education. Similarly, the incidence of perinatal and maternal mortality from DKA during pregnancy has decreased in the past several decades.48,49 As is true for nonpregnant patients, DKA during pregnancy occurs predominantly in patients with type 1 DM. The higher risk for DKA during pregnancy reflects the metabolic adaptations of pregnancy, including peripheral insulin resistance.28
During pregnancy, DKA occurs most commonly during the second and third trimesters.50 It is associated with (1) emesis, (2) infection, (3) poor compliance or noncompliance, (4) insulin pump failure, (5) use of beta-adrenergic receptor agonists, (6) use of corticosteroids, and (7) poor medical management.48 The infection rate in pregnant women with pregestational type 1 DM is 3.2 times higher than that in nondiabetic pregnant women.51 DKA may be the first clinical sign of type 1 DM during pregnancy.52,53 Beta-adrenergic receptor agonists, which are used to treat preterm labor, and corticosteroids, which are used to accelerate fetal lung maturity, both have counterregulatory pharmacologic effects that oppose insulin action. Beta-adrenergic agonist tocolytic therapy, with or without concurrent corticosteroid therapy, and by any route of administration, can precipitate DKA during pregnancy.54 Beta-adrenergic receptor stimulation worsens glucose intolerance by stimulating glucagon secretion55; beta-adrenergic receptor agonists may be well tolerated in pregnant women with DM if higher insulin requirements are anticipated and doses are adjusted in response to frequent blood glucose determinations.53,54
Nonreassuring fetal heart rate patterns during episodes of maternal DKA have been described.56 After appropriate medical management of maternal DKA, preterm uterine contractions stopped and fetal heart rate patterns normalized. The mechanism of fetal compromise during DKA is unclear, but it may be related to changes in uterine blood flow. Blechner et al.57 demonstrated that uterine artery blood flow is reduced by acute maternal metabolic acidosis. A single case report demonstrated reversible redistribution of fetal blood flow during an episode of maternal DKA on the basis of Doppler pulsatility indices of the umbilical and middle cerebral arteries.58
There are three case reports of HNS during pregnancy.59–61 No conclusion can be drawn about HNS and pregnancy, except that HNS rarely occurs during pregnancy.
Hypoglycemia.
Hypoglycemia is a significant health risk for pregnant women with pregestational type 1 DM, occurring in 33% to 71% of these patients.62–65 This rate is 3 to 15 times higher than that in similar groups of nonpregnant patients with type 1 DM62,63; 80% to 84% of severe hypoglycemia episodes occur before 20 weeks’ gestation.64,65 In one study, patients with pregestational type 2 DM or gestational DM requiring insulin therapy experienced no episodes of severe hypoglycemia.63 The risk for hypoglycemia during pregnancy in patients with type 1 DM increases with tight glucose control.62,64 This pattern mirrors the clinical experience in nonpregnant women with type 1 DM, in which a threefold rise in the occurrence of severe hypoglycemia results from tight insulin control.66 In both pregnant and nonpregnant patients with type 1 DM, counterregulatory hormone responses to hypoglycemia are impaired after intensive insulin therapy.67,68 Two small series suggest that acute mild to moderate maternal hypoglycemia is not associated with acute alterations in fetal well-being in pregnant women with type 1 DM.67,69
Other Complications.
The relationship between pregnancy and the development of macrovascular complications of DM is largely unknown. Patients with pregestational type 1 DM have higher systolic and diastolic blood pressures during pregnancy, and they are three times more likely than nondiabetic control subjects to have gestational hypertension.70,71 In women with pregestational type 1 DM, the risk for preeclampsia is increased with increased severity of diabetes (White classification), and proteinuria early in pregnancy is associated with an increased risk for adverse outcomes.72 Myocardial infarction is a rare complication.73 The effect of gestational hypertension on the progression of atherosclerotic disease in diabetic patients is unclear.
Pregnancy may accelerate the development of proliferative retinopathy, a microvascular complication of DM. Hyperglycemia and hypertension are also associated with the progression of retinopathy.74,75 The onset of strict glycemic control may transiently exacerbate diabetic retinopathy in both pregnant and nonpregnant patients with type 1 DM. The Diabetes Control and Complications Trial demonstrated that strict glycemic control is justified in nonpregnant patients.16
In contrast to diabetic retinopathy, pregnancy does not accelerate the progression of diabetic nephropathy.76 It is unclear whether pregnancy accelerates the progression of somatic or autonomic neuropathy in diabetic women.
How Does Diabetes Mellitus Affect the Mother and Fetus?
Both pregestational and gestational DM are associated with higher rates of gestational hypertension, polyhydramnios, and cesarean delivery.47,76–78 The incidence of cesarean delivery is higher in women with pregestational DM than in women with gestational DM.47,76,78 Trial of labor after cesarean delivery (TOLAC) in patients with gestational DM is associated with rates of operative vaginal delivery and repeat cesarean delivery that are higher than those found in nondiabetic controls.79 Pregestational DM—but not gestational DM—is associated with a twofold to threefold increase in the incidence of preterm labor and delivery.76,80
Box 43-4 lists the fetal complications of maternal DM during pregnancy. Fetal macrosomia is a well-recognized complication of maternal DM. Most studies suggest that both pregestational DM and gestational DM result in an increased incidence of fetal macrosomia.81–83 Depending on the definition of macrosomia (4000 g versus 4500 g), pregestational DM results in fetal macrosomia in 9% to 25% of women—a fourfold to sixfold higher rate than in nondiabetic controls.
Macrosomia results in an increased risk for shoulder dystocia and birth trauma with vaginal delivery.77,84,85 Moreover, when comparisons are made within birth weight categories above 4000 g, pregnancies in diabetic women have a higher risk for shoulder dystocia than nondiabetic women.86 The use of intensive insulin therapy may reduce the risk for birth trauma in women with pregestational DM.87 Several mechanisms have been suggested for the development of fetal macrosomia in diabetic pregnancy. Maternal hyperglycemia can result in fetal hyperglycemia, with reactive fetal hyperinsulinemia and an anabolic response in the fetus.88 Shoulder dystocia may reflect the excessive growth of the fetal trunk (relative to the fetal head) in response to fetal hyperinsulinemia.89
Women with pregestational DM are at increased risk for fetal anomalies (see Box 43-4). The incidence of major anomalies, estimated to be 6% to 10%, is five times higher than in nondiabetic controls.47,90–92 Overall, cardiovascular anomalies are most common, followed by anomalies of the central nervous system (CNS). The caudal regression syndrome is uncommon, but it is 200 times more likely in diabetic than in nondiabetic pregnancies.90 The incidence of major congenital anomalies in infants of women with gestational DM is 3% to 8%, which is lower than in infants of women with pregestational DM.76
Metabolic factors that may be involved in the development of fetal structural malformations in diabetic pregnancies include hyperglycemia, hypoglycemia, arachidonic acid, polyol pathways, mitochondrial dysfunction, and apoptosis.92 Most fetal structural malformations that occur during diabetic pregnancies are likely to have a multifactorial etiology. However, hyperglycemia during the period of critical organogenesis before the seventh week after conception is probably the single strongest etiologic factor in diabetic women and may be associated with embryonic oxidative stress.92,93
Studies have suggested that patient education and strict glycemic control during the preconception period may reduce the rate of major congenital anomalies from 10% to 1% in patients with pregestational DM.94 The latter figure is similar to the baseline risk for major structural malformations in the general population. Strict glycemic control initiated during the preconception period also increases the incidence of maternal hypoglycemic episodes. These studies suggest that hypoglycemia is not a significant factor in the etiology of human malformations, because the rate of anomalies decreased 10-fold despite hypoglycemic episodes.94 Similarly, strict glycemic control before conception also has been associated with a threefold decrease in the incidence of spontaneous abortion in women with pregestational DM.95 Dicker et al.96 observed normal induced ovulation, in vitro fertilization, and early embryonic development in a small series of infertile patients with pregestational DM who attended a preconception diabetes clinic. However, only 36% of women with known pregestational DM receive appropriate medical care before conception.
During the 1950s to 1970s, the perinatal mortality rate in women with pregestational DM was 15% to 18%.47 Subsequent studies noted a decrease to 2%, a rate similar to that in nondiabetic controls.76 In contrast, one study noted a rate of 8%, three times greater than in nondiabetic controls.83 If the entire population is considered, the perinatal mortality rate likely remains higher in patients with pregestational DM than in nondiabetic controls. The rate in patients with gestational DM is intermediate between the rate in women with pregestational diabetes and the rate in nondiabetic controls.76,83
Historically, intrauterine fetal death was responsible for approximately 40% of the perinatal deaths in women with DM; 68% of the stillbirths occurred between 36 and 40 weeks’ gestation.45,83 In contemporary reports, the ratio of intrauterine deaths to neonatal deaths in diabetic pregnancies has varied from 0 to 1.0. Fetal macrosomia is a risk factor for intrauterine fetal demise in both diabetic and nondiabetic pregnancies. Recurrent episodes of intrauterine hypoxia can occur in diabetic pregnancies that end in stillbirth; episodes of hypoxia may reflect reduced uteroplacental blood flow and changes in fetal carbohydrate metabolism. Congenital anomalies have now emerged as the leading cause of perinatal mortality in diabetic pregnancies.92 This change likely reflects better obstetric care during pregnancy, despite the lack of adequate glycemic control before conception.
Two series that involved women who delivered between 1950 and 1979 demonstrated an incidence of neonatal respiratory distress syndrome (RDS) in diabetic pregnancies that was 6 to 23 times that in nondiabetic controls.47,97 Respiratory distress is more common among newborns who are delivered preterm or who are surgically delivered without labor. Later studies of patients with both pregestational and gestational DM have not demonstrated a significant difference in the incidence of neonatal RDS between diabetic and nondiabetic pregnancies.76,98,99
The level of glycemic control during pregnancy affects the amniotic fluid phospholipid profile. In pregnancies of patients with poorly controlled diabetes, there may be a higher incidence of immature amniotic fluid fetal lung profiles at 34 to 38 weeks’ gestation without an increase in the rate of clinical respiratory distress syndrome.99,100 In reliably dated pregnancies of diabetic patients, fetal lung maturity testing has little clinical benefit.101
Neonatal hypoglycemia occurs in 5% to 12% of cases of pregestational and gestational DM.76 This represents a 6-fold to 16-fold higher risk for neonatal hypoglycemia than in nondiabetic controls. Neonatal hypoglycemia likely results from sustained fetal hyperinsulinemia in response to chronic intrauterine hyperglycemia. Clinical studies have demonstrated higher fetal insulin levels and exaggerated fetal insulin responses to acute maternal hyperglycemia in diabetic pregnancies.102,103 An acute increase in maternal glucose concentration, as might occur if a dextrose-containing solution was used for intravenous hydration during administration of neuraxial anesthesia, can lead to reactive neonatal hypoglycemia, even in nondiabetic women.104
There is a twofold to fivefold higher incidence of neonatal hyperbilirubinemia in women with pregestational and gestational DM than in nondiabetic controls.76 Other associated factors include the severity of gestational DM and excess maternal weight gain during pregnancy.105,106 Both the etiology and the clinical significance of neonatal hyperbilirubinemia are unknown, although one study noted the absence of long-term morbidity.76
Offspring of diabetic mothers are at increased risk for development of DM, likely from a combination of genetic and intrauterine environmental factors. Despite the well-known association of type 1 DM with human leukocyte antigen markers, studies of monozygotic human twins have suggested that genetic factors have a greater role in type 2 DM than in type 1 DM (100% versus 20% to 50% concordance, respectively).105 In addition, fathers with type 1 DM are five times more likely than mothers with the same disease to have a child with type 1 DM. The intrauterine environment also affects the development of glucose intolerance in offspring.31
Some investigators have suggested that cognitive development may be impaired in the children of diabetic mothers,107 but this issue remains controversial.
Obstetric Management
Glycemic Control
Early, strict glycemic control is the best way to prevent fetal structural malformations in women with pregestational DM.92,94 Determination of hemoglobin A1c concentrations may help the physician determine the adequacy of preconceptional glycemic control.
During pregnancy, the patient should frequently determine capillary blood glucose concentration using a reflectance meter.108 Continuous glucose monitoring systems (e.g., transdermal, subcutaneous) are more recent approaches.109 Glucose determinations guide adjustments in diet and insulin therapy. In general, insulin requirements increase progressively during the second and third trimesters. Both maternal and perinatal outcomes seem to improve when maternal glycemic control approaches that observed in normal pregnancies. Opinions vary about the optimum target glucose concentration in patients with pregestational DM, but a fasting blood glucose concentration of 60 to 95 mg/dL seems appropriate. Of course, strict glycemic control increases the risk for maternal hypoglycemia.
Therapeutic insulin is available in several forms. Initially, insulin was isolated as a natural product from domestic animals (e.g., cattle, pigs). In the past 20 years, synthetic human insulin has become commercially available and has largely replaced beef and pork insulin in human medicine, with an expected decrease in immune reactions among human recipients.110
The goal of insulin therapy is to provide plasma insulin concentrations that lead to tight glucose control without hypoglycemia. This goal is facilitated by the availability of several insulin preparations with different subcutaneous absorption rates (Table 43-2).109 Regular insulin can be administered by the intravenous or subcutaneous route. Regular insulin administered intravenously has a half-life of approximately 4 minutes.111 Other native insulins listed in Table 43-2 (i.e., neutral protamine Hagedorn [NPH], the zinc suspensions lente and ultralente) represent chemical complexes of regular insulin with protamine or zinc; subcutaneous administration of these insulins is associated with slower absorption and onset of action. Lente and ultralente insulins have been replaced clinically by insulin analogues and are of only historical interest. An alternative therapeutic strategy is to administer a rapid-acting insulin by the subcutaneous route using a continuous programmable pump.112 It is unclear whether continuous subcutaneous pump–administered insulin is clinically superior to intermittent subcutaneous injections of currently available insulins.113,114
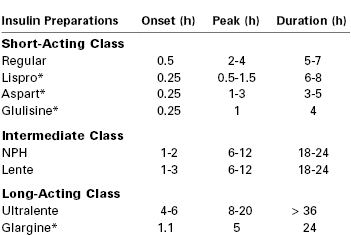
Human insulin therapy has fundamentally changed in recent years through the development of insulin analogues.115,116 These molecules have specific chemical substitutions in portions of the human insulin protein not involved in receptor binding. Both short-acting and long-acting insulin analogues are in clinical use. Lispro and aspart are rapid acting, with a more physiologic onset and offset than regular insulin. Glargine is relatively insoluble at neutral pH in the subcutaneous compartment. In contrast to ultralente insulin, subcutaneous glargine has a sustained release without an initial peak of activity. Lispro, aspart, and glargine have all been used safely during human pregnancy.
Because insulin requirements decrease abruptly at delivery, it is important to verify the times, doses, insulin preparations, and routes of administration in the 24 hours before delivery to avoid maternal postpartum hypoglycemia.
Management of DKA is similar in pregnant and nonpregnant women. It involves (1) intravenous hydration, (2) intravenous insulin, (3) treatment of the underlying cause of DKA, (4) careful monitoring of blood glucose and electrolyte levels, and (5) restriction of bicarbonate therapy to cases of extreme acidosis.9,10,48 In addition, left uterine displacement should be maintained and supplemental oxygen should be administered. Initial management of the critically ill pregnant woman should focus on the effective management of DKA. Fetal compromise is likely to resolve with appropriate medical management.56,57
Diet and exercise are the initial therapeutic approaches for glycemic control in women with gestational DM. Insulin therapy is initiated if the fasting glucose measurement exceeds a threshold of 80 to 105 mg/dL.23,117 In the past, oral hypoglycemic agents were not used extensively in pregnancy, primarily because of concerns about potential teratogenicity and fetal hyperinsulinemia. In current practice, many women with gestational DM are treated with glyburide, glipizide, or metformin.118,119 Concerns about the long-term health implications of gestational diabetes have resulted in national initiatives for postpartum metabolic surveillance.120,121 The goal is to identify and treat postpartum type 2 DM, with an emphasis on lifestyle interventions.
Timing of Delivery
Timing of delivery is important in the management of diabetic pregnancies. White46 noted, “Our problem must [be] … to prevent premature delivery of the infant of the diabetic mother prior to the period of its viability … and, secondly, the termination of the pregnancy at the point of viability and before the dreaded late intrauterine accident can occur.” Typically a nonstress test is performed twice weekly in patients with pregestational DM, beginning at 32 weeks’ gestation.122,123 A nonreactive nonstress test should prompt the performance of a fetal biophysical profile (see Chapter 6). Risk factors for abnormal fetal testing in diabetic pregnancies include maternal nephropathy, hypertension, and poor glycemic control.124 No consensus exists regarding antepartum testing in women with well-controlled gestational DM.23 Patients with poorly controlled gestational DM should probably undergo antepartum fetal surveillance similar to that in patients with pregestational DM.23,117
In the presence of reassuring fetal testing, delivery can be delayed until after 38 weeks’ gestation.122 If fetal testing is abnormal and amniotic fluid analysis indicates fetal pulmonary maturity, the fetus should be delivered as soon as possible. If fetal testing is abnormal but amniotic fluid analysis suggests that the fetal lungs are immature, decisions about the timing of delivery are more difficult.
The decision regarding the method of delivery requires consideration of estimated fetal weight, fetal condition, cervical dilation and effacement, and previous obstetric history. The obstetrician may choose elective cesarean delivery in the diabetic parturient with evidence of fetal macrosomia to decrease the risk for shoulder dystocia.
Anesthetic Management
Few studies exist concerning the anesthetic management of pregnant women with DM. In general, neuraxial analgesia is the preferred technique for labor and cesarean delivery, but clinical decisions about these patients must be guided by logical extensions of studies of nonpregnant diabetic patients and nondiabetic pregnant patients.
Preanesthetic evaluation of the woman with DM should include a history and physical examination that focuses on the identification of the acute and chronic complications of DM (see Box 43-1). There are no published data on the relationship between the complications of DM and responses to anesthetic agents or on anesthetic outcomes in pregnant patients. In a study of nonpregnant diabetic patients, preoperative evidence of autonomic cardiovascular dysfunction was predictive of the need for a vasopressor during general anesthesia.125 Because of the potential for hypotension during neuraxial anesthesia, noninvasive testing of autonomic function may be useful in obstetric patients with pregestational DM. For example, in nonpregnant diabetic patients the corrected QT interval on an electrocardiogram correlates with the severity of autonomic neuropathy.126 Patients with evidence of autonomic dysfunction may benefit from more frequent blood pressure determinations and more vigorous intravenous hydration before and during the administration of neuraxial anesthesia. Gastroparesis is a manifestation of autonomic neuropathy in diabetic patients.127 In nonpregnant diabetic patients, autonomic neuropathy is associated with a decreased cough reflex threshold and a higher incidence of obstructive sleep apnea.128,129
Several studies have examined the maternal, fetal, and neonatal effects of neuraxial anesthesia for cesarean delivery for women with pregestational DM.130–133 Datta and Brown130 observed that spinal anesthesia was associated with a slightly but significantly lower umbilical cord blood pH measurement at delivery in patients with pregestational DM than in similar patients who received general anesthesia for cesarean delivery. Subsequently, these investigators noted an association between fetal acidosis and peripartum maternal hypotension in patients with pregestational DM who received epidural anesthesia for cesarean delivery.131 In both studies, acute maternal hyperglycemia—secondary to intravenous hydration with 5% dextrose before administration of neuraxial anesthesia—was a potentially confounding factor.130
Neonatal acidosis is not likely to occur during spinal or epidural anesthesia for cesarean delivery in diabetic parturients, provided that (1) maternal glycemic control is satisfactory, (2) the patient receives aggressive preanesthetic volume expansion with a non–dextrose-containing balanced salt solution, and (3) hypotension is treated promptly and aggressively.132,133
Thalme and Engstrom134 demonstrated normal umbilical arterial blood pH measurements after the administration of general anesthesia in a small series of patients with pregestational DM.
After administration of epidural anesthesia for cesarean delivery, Ramanathan et al.133 observed an increased incidence of neonatal hypoglycemia in patients with pregestational DM compared with nondiabetic controls (35% versus 7%, respectively). In this study, maternal glycemic control was fair (mean fasting plasma glucose level was 127 mg/dL), a non–dextrose-containing solution was used for intravenous hydration, and intravenous insulin therapy was adjusted on the basis of frequent blood glucose determinations. This study illustrates the neonate’s vulnerability to hypoglycemia after a diabetic pregnancy despite meticulous anesthesia care at the time of delivery.
A single case report describes a parturient who received combined spinal-epidural labor analgesia and subsequently became hypoglycemic. The authors hypothesize that the rapid decrease in catecholamine levels from the resultant analgesia led to hypoglycemia.135
Maternal insulin requirements decrease with the onset of labor, increase again during the second stage of labor, and decrease markedly during the early postpartum period.40,41 Intravenous insulin therapy is the most flexible method of treatment during this period of rapid change. Absorption of subcutaneous insulin may be unpredictable and may increase the risk for maternal hypoglycemia, especially during the postpartum period.41 Moreover, strict glycemic control in pregnant women with type 1 DM increases the risk for maternal hypoglycemia as a result of impaired counterregulatory hormone responses (as discussed earlier).
Intravenous glucose and insulin infusions during the peripartum period should be titrated to maintain a maternal blood glucose concentration of 70 to 90 mg/dL. During active labor, the glucose requirement is 2.5 mg/kg/min or more.38 For cesarean delivery with patients using subcutaneous insulin pumps, a preoperative strategy should be formulated for perioperative insulin pump management. This plan should identify the individual(s), other than the patient, who are experienced and knowledgeable in adjusting her insulin pump if the patient is not able to adjust it herself during surgery. Some patients and obstetricians prefer discontinuing the subcutaneous insulin pump preoperatively in favor of an intravenous insulin infusion. The preanesthetic evaluation is an excellent opportunity to discuss with the patient the expected changes in insulin requirements at the time of delivery. The preprocedure and postprocedure “time-outs” are excellent opportunities to discuss the plan for perioperative pump management and other diabetic management concerns with all team members.
Many perioperative strategies have been proposed for metabolic control in nonpregnant patients with DM.136–138 No convincing evidence suggests that one clinical strategy for perioperative diabetic control is superior in terms of patient outcome. Frequent blood glucose measurements (e.g., at 30- to 60-minute intervals), followed by appropriate adjustment of glucose and insulin infusions, represent the cornerstone of optimal perioperative care in patients with DM.
There are no published data on the effects of DM on the pharmacokinetics and pharmacodynamics of anesthetic agents in pregnant women. In nonpregnant women, DM is associated with (1) a delayed onset of muscle relaxation with tubocurarine and (2) prolonged blockade with vecuronium.139,140
The diabetic stiff-joint syndrome has been associated with difficult direct laryngoscopy and tracheal intubation in patients with DM.141,142 This syndrome occurs in patients with long-standing DM type 1 and is associated with nonfamilial short stature, joint contractures, and tight skin.143 Limited movement of the atlanto-occipital joint may result in difficult direct laryngoscopy and tracheal intubation. During the preanesthesia evaluation of patients with DM, the anesthesia provider can screen for the stiff-joint syndrome by looking for the “prayer sign” (Figure 43-4). Management is controversial. Some authorities recommend preanesthesia flexion-extension radiographic studies of the cervical spine followed by awake tracheal intubation.142 Others have expressed doubt about the clinical significance of this syndrome and the reported frequency of airway management problems.144,145 The term diabetic scleredema is synonymous with stiff-joint syndrome. There is one case report of a pregnant patient with pregestational DM and diabetic scleredema who experienced anterior spinal artery syndrome after the administration of epidural anesthesia for cesarean delivery.146 The author suggested that spinal cord vascular compression resulted from a combination of (1) preexisting microvascular disease, (2) an epidural space that was stiff because of connective tissue disease, and (3) administration of a large volume (i.e., 35 mL) of the local anesthetic agent. In patients with a history and physical examination that suggest diabetic stiff-joint syndrome, the anesthesia provider should consider two potential problems: (1) difficult direct laryngoscopy and tracheal intubation and (2) a noncompliant epidural space.
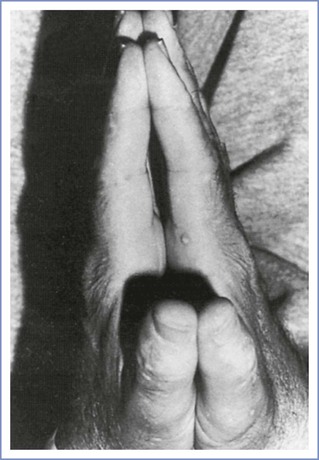
FIGURE 43-4 Inability to approximate the palmar surfaces of the phalangeal joints despite maximal effort, secondary to diabetic stiff-joint syndrome. (From Hogan K, Rusy D, Springman SR. Difficult laryngoscopy and diabetes mellitus. Anesth Analg 1988; 67:1162-5.)
Infection is an important cause of morbidity in pregnant women with pregestational DM.49 There are no published data regarding the incidence of CNS infection after the administration of neuraxial anesthesia in pregnant diabetic patients except for one case of fungal meningitis due to contaminated spinal anesthesia equipment.147 Strict aseptic technique always should be used during the administration of neuraxial anesthesia.
Thyroid Disorders
Thyroid Hormone Physiology
The follicular cells of the thyroid gland sequester iodine and synthesize thyroglobulin, an iodinated precursor protein. Thyroglobulin is secreted into the lumen of the microscopic thyroid follicles before it undergoes reuptake, proteolysis, and transfer to lysosomes, where it undergoes degradation.148 This process results in the systemic release of the thyroid hormones: thyroxine (T4) and 3,5,3′-triiodothyronine (T3). Reverse T3 (3,3′,5′-triiodothyronine) is a structural variant with much less physiologic potency in most target organs.149
Thyroid hormone synthesis and release are controlled primarily by thyroid-stimulating hormone (TSH)—a trophic hormone from the pituitary—and the supply of iodine. The thyroid hormones normally participate in a negative feedback loop that regulates TSH secretion (Figure 43-5) and thyrotropin-releasing hormone production in the hypothalamus.150
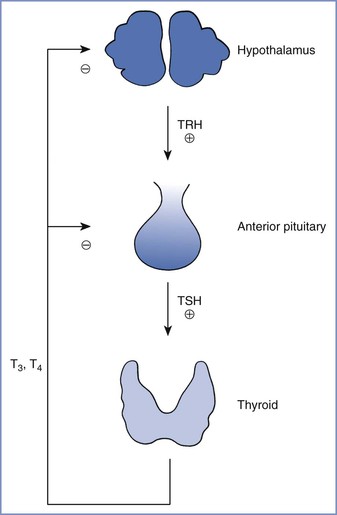
FIGURE 43-5 Normal feedback control of thyroid hormone secretion. TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, thyroxine. (From Davies PH, Franklyn JA. The effects of drugs on tests of thyroid function. Eur J Clin Pharmacol 1991; 40:439-51.)
Thyroid hormones are highly bound to protein in the blood. In euthyroid nonpregnant humans, the normal total serum concentrations of T4 and T3 are 50 to 150 nmol/L and 1.4 to 3.2 nmol/L, respectively.151 The unbound or free fractions of T4 and T3 are 0.03% and 0.3% of total circulating T4 and T3, respectively.152 Similar proportions of T4 and T3 are distributed among the three major plasma proteins that bind thyroid hormones, which are (1) thyroxine-binding globulin (70% to 80%), (2) thyroxine-binding prealbumin or transthyretin (10% to 20%), and (3) albumin (10% to 15%).153,154 The serum concentration of unbound or free T4 is typically the major determinant of thyroid hormone activity in target tissues. Thyroid hormones are temporarily inert while bound to plasma proteins. Changes in the concentrations of thyroxine-binding proteins can occur during various physiologic states (e.g., pregnancy) and disease processes. Thyroid hormone action does not change with fluctuations in the total concentration of T4 as long as the concentration of free T4 remains constant.
Thyroid hormone is an endocrine regulator in many target organs (e.g., liver, kidneys, skeletal and cardiac muscles, brain, pituitary, placenta).155 The defined physiologic effects of thyroid hormones are mediated by regulation of specific gene products. These effects include (1) somatic and nervous system development, (2) calorigenesis, (3) augmented skeletal and cardiac muscle performance, (4) intermediary metabolism, and (5) feedback control.156
In target tissues, the molecular actions of T4 begin with the enzymatic deiodination of T4 to T3. Iodothyronine deiodinase is widely distributed in the body and occurs in three molecular forms.157 Only 20% of the daily T3 production is secreted by the thyroid gland; the rest is formed by peripheral deiodination.158 In the classic model of thyroid hormone action, T3 enters the nuclei of target cells, binds to specific thyroid hormone receptors, and alters genomic transcription of specific proteins.159 Research has now characterized other mechanisms of thyroid hormone action, including mitochondrial transcription and cytoplasmic or cell-surface nontranscriptional effects.160,161 The thyroid hormone receptor belongs to a family of structurally related, intracellular ligand-binding proteins.160 Variations in the number and types of thyroid hormone receptors, as well as receptor linkage to development- or tissue-specific genomic expressions, provide additional levels of physiologic control and vulnerability to disease processes.157
Antithyroid medications may affect single or multiple steps in thyroid hormone synthesis and release, as well as concentrations of plasma binding proteins, deiodinase activity, and peripheral uptake of thyroid hormones.153,162,163
Laboratory evaluation of thyroid function consists of two measurements. First, the serum concentration of free T4 can be directly measured or indirectly calculated. Second, the serum concentration of TSH is measured to assess the negative feedback loop that controls the thyroid gland. The TSH concentration is judged as appropriate or inappropriate in the context of the serum concentration of free T4.
During normal human pregnancy, the serum concentration of thyroxine-binding globulin (TBG) steadily increases until it reaches a plateau at 20 weeks’ gestation, when it is 50% greater than the nonpregnant level.151 The greater concentration of TBG results from a prolonged half-life—not higher synthesis—during pregnancy.154 The normal pregnant woman is euthyroid because the serum concentrations of free T4 and T3 are in the normal or low-normal range for nonpregnant humans.151 However, the increased concentration of TBG means that total serum concentrations of T4 and T3 during pregnancy are at or above the upper limit of normal for nonpregnant women.151,164
Human chorionic gonadotropin (hCG) is a placental protein that shares some structural features with TSH. The serum concentrations of TSH and hCG have an inverse relationship during normal human pregnancy,151 reflecting the mild TSH-like activity that results from increased plasma concentrations of hCG during early pregnancy.165,166
Maternal iodine availability is decreased during pregnancy because of greater fetal uptake and increased maternal renal clearance.167 In geographic areas with marginal iodine supplies, the lower availability may predispose the mother to goiter unless she receives dietary iodine supplementation.151,168,169
Hyperthyroidism
Definition and Epidemiology
Hyperthyroidism is defined as an abnormal increase in the serum concentration of unbound or free thyroid hormones. The prevalence of hyperthyroidism in the general population is 0.2% to 1.9%, with a female-to-male ratio of 10 : 1.170,171 The etiology of hyperthyroidism is listed in Box 43-5. Graves’ disease is responsible for 70% to 90% of cases; thyroiditis and the combined category of toxic adenoma and toxic multinodular goiter each account for approximately 5% of cases. There are multiple levels of interaction between the thyroid and reproductive endocrine systems in women, with specific implications for patients with hyperthyroidism and hypothyroidism.172
Pathophysiology
Graves’ disease is an autoimmune thyroid disease.170,173 Its etiology is likely multifactorial and includes both environmental (e.g., stress, hormones) and genetic influences. Several autoantibodies against thyroid tissue have been described in patients with this disease. Autoantibodies directed against the TSH receptor in the thyroid gland may either augment or inhibit TSH action, depending on their binding specificities. These antibodies are called thyroid receptor antibodies (TRAbs). The binding specificities of TRAbs in the blood of each patient with Graves’ disease affect the net thyroid-stimulating activity. Autoantibodies against thyroid peroxidase, the sodium-iodine cotransporter, and thyroglobulin also have been described in patients with Graves’ disease.
Among untreated patients with Graves’ disease, approximately 20% undergo spontaneous remission.173 However, the prognosis for individual patients cannot be predicted from results of clinical or laboratory examinations.
Clinical Presentation and Diagnosis
Hyperthyroidism presents as a physiologic state dominated by an increased metabolic rate. A hyperthyroid symptom scale has been developed on the basis of the following 10 clinical factors: nervousness, sweating, heat intolerance, hyperactivity, tremor, weakness, hyperdynamic precordium, diarrhea, appetite, and level of incapacitation.174 This symptom scale has been useful to follow the clinical course of patients with Graves’ disease. Exophthalmos or infiltrative ophthalmopathy is clinically apparent in most patients.173–175 Other physical signs may occur at low frequency, including pretibial myxedema or dermopathy (1% to 2%) and nail changes or acropachy (< 1%). The infiltrative ophthalmopathy in Graves’ disease is caused by enlargement of both the extraocular muscle bodies and intraorbital adipose tissue. The pathogenetic mechanism involves abnormal accumulation of hyaluronic acid and edema within these tissues; the orbital fibroblast appears to be the primary target cell of this autoimmune process.175
Hyperthyroidism stimulates the cardiovascular system in excess of the underlying increased metabolic rate, resulting in a hyperkinetic circulatory state.163,176 Myocardial contractility, heart rate, stroke volume, and ventricular size all increase, and peripheral vascular resistance decreases in skin and muscle. Thyroid hormones can affect the ratio of alpha- and beta-adrenergic receptors in the heart.176 Cardiomyopathy can be demonstrated during exercise in hyperthyroid patients, independent of beta-adrenergic receptors; it is reversible with normalization of thyroid function.177
The diagnosis of hyperthyroidism depends on increased serum concentrations of unbound or free T4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


