Disorders of Sulfur-Containing Amino Acid Metabolism
Bernd Christian Schwahn
Sulfur-containing amino acids have various roles: They mediate the transfer of methyl groups for virtually all transmethylation reactions; they provide reactive thiol groups that are needed for detoxification of endogenous and exogenous substances; they help maintain the intracellular redox potential; and they are a source of sulphate. Thirteen disorders of sulphur amino acid metabolism have been consistently described, 11 of them potentially leading to disease.
HOMOCYSTINURIA
Severe hyperhomocysteinemia, defined as plasma total homocysteine (tHcy) concentrations above 100 μmol/L, is generally caused by single-enzyme deficiencies of homocysteine metabolism. When concentrations of free homocysteine exceed the plasma protein binding capacity, the disulfide homocystine forms nonenzymatically and is excreted in urine, hence causing homocystinuria. The term homocystinuria is sometimes used to indicate the most common form of the disease, which is caused by defective activity of the enzyme cystathionine β-synthase (CBS).1 Homocystinuria, however, results from a defect in CBS and from defects in the folateand cobalamin-dependent remethylation cycle (see Chapter 147). 
CLASSICAL HOMOCYSTINURIA (CBS DEFICIENCY)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Classical homocystinuria presents as multisystemic disease with a dysplasia of connective tissue, with a predisposition to arterial and venous thromboembolism, and with mental retardation. Clinical variability is wide,2 but presentation is usually in the first decade with the exception of embolism, which occurs later. Homocystinuria is one of the few disorders of amino acid metabolism in which clinical manifestations tend to be progressive in adulthood, because many clinical manifestations result from arteriosclerosis and thrombotic complications.
The most characteristic feature of this disorder is subluxation of the ocular lens, which occurs in almost all untreated individuals until adulthood. Most patients have osteoporosis and skeletal abnormalities similar to those seen in Marfan syndrome, such as tall stature, scoliosis, genu valgum, pes cavus, arachnodactyly, and pectus carinatum or excavatum.3 In homocystinuria, however, the joints tend to be limited in mobility rather than hypermobile. Mental retardation is common, although it is often mild, and many individuals have psychiatric disturbances.4
 METABOLIC DERANGEMENT, PATHOPHYSIOLOGY
METABOLIC DERANGEMENT, PATHOPHYSIOLOGY
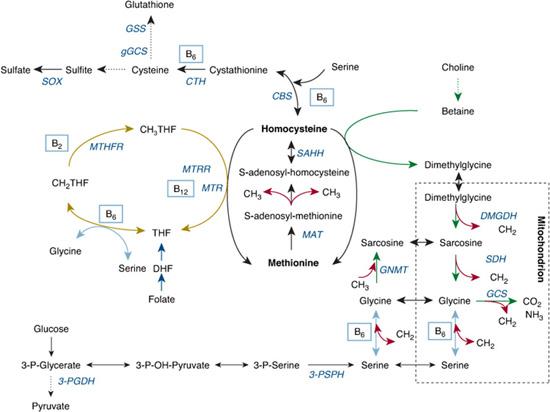
The disturbance in metabolism resulting in homocystinuria is shown in Figure 138-1. CBS initiates the first step of homocysteine elimination. As a consequence of CBS deficiency, homocysteine, SAH, SAM, and methionine accumulate when methionine intake exceeds the residual transsulfuration and total remethylation activity. Moreover, high SAH inhibits many transmethylases, which increases accumulation of SAM and methionine. Increased homocysteine facilitates remethylation, which leads to further accumulation of methionine. The intermediate metabolites cystathionine and cysteine are decreased. 
 GENETICS
GENETICS
CBS deficiency (OMIM No. 236200) is an autosomal recessive trait. The CBS gene has been mapped to chromosome 21q22.3, and more than 100 different mutations have been identified, most of them being private.6,7 However, two mutations, I278T and G307S, account for 25% to 50% of all affected alleles, depending on the population. A few other mutations, including I278T, are usually associated with pyridoxine responsiveness. Antenatal diagnosis has been performed by assaying CBS activity in cultured amniocytes or cultured chorionic villi. Mutational analysis can be used for prenatal diagnosis once the mutation of the index case is known. The real incidence of CBS deficiency is not known but may vary between 1:20,000 and 1:200,000.8
 DIAGNOSTIC TESTS, DIFFERENTIAL DIAGNOSES
DIAGNOSTIC TESTS, DIFFERENTIAL DIAGNOSES 
Once hyperhomocysteinemia has been identified, usually in the range of 80 to 300 μmol/L, plasma amino acid chromatography can be used to search for hypermethioninemia and low cystine, cystathionine, and serine concentrations. Defects of folate- and cobalamin-dependent remethylation can be differentiated by a low or normal plasma methionine concentration and increased cystathionine. Increased urinary methylmalonic acid excretion and megaloblastic anemia suggest either concomitant cobalamin deficiency or a defect in cobalamin transport or metabolism. Severe folate or cobalamin deficiency should be ruled out by measuring serum vitamin concentrations (see Table 138-1 and Chapter 147). The diagnosis can be confirmed by measuring enzyme activity in fibroblasts or in phytohemagglutinin-stimulated lymphocytes.
 TREATMENT, PROGNOSIS, AND LONG-TERM OUTCOME
TREATMENT, PROGNOSIS, AND LONG-TERM OUTCOME
Approximately half of all CBS-deficient individuals respond with clear biochemical improvement to supplementation with large doses of pyridoxine.11 Responsiveness is determined by the properties of the mutant apoenzyme and should be tested in every patient by administration of pyridoxine for at least a week (500 mg daily in children and 1000 mg per day in adults). Medical treatment aims to control the biochemical abnormalities to prevent complications or halt the progression of symptoms. Early treatment and maintenance of tHcy below 50 μmol/L prevents the patient’s exposure to excessive concentrations of free homocysteine and appears to be associated with a near-normal risk for thromboembolic events, improvement in behavior and IQ, and avoidance of lens dislocation.1 Those who do not respond sufficiently to pyridoxine supplementation need to be treated with a diet low in methionine and supplemented with L-cystine. In addition, the methyl donor betaine should be added to enhance remethylation. Individuals who do not restrict their methionine intake may develop extreme hypermethioninemia with betaine and are at risk for brain edema if methionine exceeds 1000 μmol/L.12
METHYLENETETRAHYDROFOLATE REDUCTASE DEFICIENCY
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Like individuals with CBS deficiency, those with 5,10-methylenetetrahydrofolate reductase (MTHFR) deficiency are at risk for arteriosclerosis and thromboembolism. They have, however, fewer skeletal symptoms, and lens dislocation is not observed.15 Most suffer from gait disturbance, a dysmyelinating encephalopathy, seizures, and mental retardation. Psychiatric disturbances are very common. The phenotype of severe MTHFR deficiency ranges from early neonatal death to nearly asymptomatic adults and is dependent on genetic and environmental modifiers. The other two remethylation defects, namely methionine synthase (MTR) deficiency and MTR reductase (MTRR) deficiency (see Table 138-2), share similarities with MTHFR deficiency, as do three other primary defects in cobalamin metabolism (see Chapter 147). These are, however, less common and are associated with megaloblastic anemia due to methylfolate trapping, which is not a feature of MTHFR deficiency.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


