Disorders of Energy Metabolism
Juan M. Pascual and Salvatore DiMauro
Surprisingly, the number of energy metabolism disorders compatible with life is still expanding, and their manifestations are reaching truly pleomorphic proportions. Collectively, these disorders spare no organ or tissue and can mimic many of the diseases routinely encountered by primary care clinicians. In addition to the well-known role of energy metabolism enzymes in balancing the flux of high-energy bonds inside cells and the supply of fuels to them, some also seem to serve multiple roles. For example, mutations in some pyruvate metabolism enzymes impair axonal migration but can also alter craniofacial configuration. Many mutations are linked to selective neuronal necrosis and apoptosis and to edema (spongio-sis) of the cerebral white matter; paradoxically, most cause enhanced excitation and epilepsy and result in increased neuronal energy demands. These unexpected manifestations probably occur because flux through energy metabolism pathways sustains the synthesis and recycling of neurotransmitters and other signaling molecules by groups of neural cells. Consequently, the brain usually bears the full burden of these diseases, but cardiac and skeletal muscles, liver, and kidney are also frequently involved.
PYRUVATE DEHYDROGENASE DEFICIENCY
Defects in the pyruvate dehydrogenase (PDH) complex are a frequent cause of lactic acidosis. PDH is a large mitochondrial matrix enzyme complex that catalyzes the oxidative decarboxylation of pyruvate to form acetyl-CoA, nicotinamide adenine dinucleotide (NADH), and CO2. Symptoms vary considerably in patients with PDH complex deficiency, and almost equal numbers of males and females are affected, despite the location of the PDH E1 alpha subunit gene (PDHA) in the X chromosome, a paradox explained by selective female X-inactivation.1 Thus, the phenotype of PDH deficiency is dictated by mutation severity (especially in males) and by the pattern of X-inactivation in females.2 Dozens of PDHA1 mutations have been identified. In addition, there are patients harboring mutations in the E1 beta subunit, the E2 dihydrolipoyl transacetylase segment of the complex, the E3 (dihydrolipoamide dehydrogenase) subunit, the E3-binding protein, the lipoyl-containing protein X, and the PDH phosphatase (eTable 159.1  ).3
).3
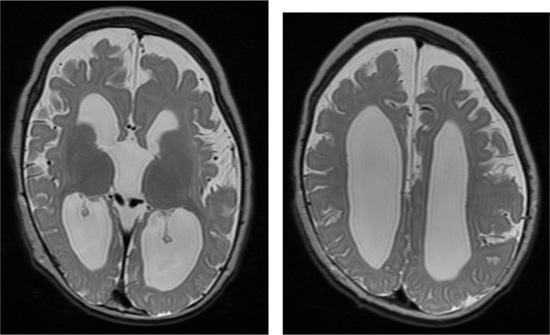
Neonates with pyruvate dehydrogenase complex (PDC) defects may present with severe aci-dosis caused by progressive lactate and pyruvate accumulation, hypotonia, microcephaly, partial or total agenesis of the corpus callosum (see Fig. 159-1), and dysmorphic features similar to those seen in fetal alcohol syndrome. The acidosis is refractory to treatment, but thiamine pyrophosphate and dichloroacetate are often administered. If these infants survive this initial phase, they have severe neurological impairment and often die by about 3 years of age. Some children present later during the first year of life with Leigh disease. The clinical manifestations can be indistinguishable from forms caused by respiratory chain disorders (see Chapter 188).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


