Congenital Liver Failure and Neonatal Hepatitis
EPIDEMIOLOGY
Neonatal hepatitis is a vague term that describes a heterogeneous group of disorders occurring in infants up to 3 months of age due to a variety of insults. It includes all forms of neonatal cholestasis not attributed to extrahepatic biliary tract obstruction. In many instances, a specific inciting event, infectious agent, or metabolic cause cannot be found. Approximately 40% of cases of neonatal cholestasis are due to neonatal hepatitis.1
Neonatal acute liver failure (ALF) is rare but often fatal. It is defined as the development of hepatic necrosis associated with hepatic encephalopathy and coagulopathy within 8 weeks of the onset of liver disease without evidence of chronic liver disease. This definition is challenging given prenatal liver disease is challenging to identify, and encephalopathy in neonates is difficult to distinguish. The Pediatric Acute Liver Failure study group defined ALF as hepatic-based coagulopathy defined as prothrombin time (PT) of 15 seconds or longer or international normalized ratio (INR) of 1.5 or more, not corrected by vitamin K, in the presence of clinical hepatic encephalopathy, or a PT of 20 or more or INR of 2 or more regardless of the presence or absence of clinical hepatic encephalopathy, along with biochemical evidence of acute liver injury and no known evidence of chronic liver disease.2
This chapter review focuses on known causes of neonatal hepatitis, concentrating on those that can progress to ALF. Please refer to the chapters on conjugated hyperbilirubinemia, congenital infections, and metabolic diseases for additional discussion.
PATHOPHYSIOLOGY
Sporadic neonatal hepatitis is often ascribed to a specific injury from infectious, ischemic, or environmental factors, whereas familial neonatal hepatitis is assumed to be caused by genetic defects in hepatic metabolic or excretory function. Idiopathic neonatal hepatitis is a generic name for indeterminate causes of ALF. Biopsies classically show formation of syncytial hepatic giant cells, variable inflammation, and lobular cholestasis. A review of 62 cases showed lobular extramedullary hematopoiesis was present in 74%, lobular cholestasis in 84%, and mild patchy chronic inflammation in 54%. Fibrosis was present in 30% of cases. Most cases (49%) remained idiopathic, but 16% of cases were diagnosed with hypopituitarism and 14% as having biliary atresia or Allagille syndrome. The biopsy findings did not readily distinguish between the specific etiologies.3
Neonatal Hepatitis
The classic pathology of neonatal hepatitis is characterized by predominantly parenchymal lobular inflammation with preservation of the zonal distribution of portal tracts and central veins. There is ballooning degeneration of hepatocytes. There can be intense giant cell transformation. Cholestatic features can also be seen, such as cytoplasmic feathery degeneration, formation of cholestatic rosettes, cholestatic plugs, and Kupffer cell activation. No obstructive pathology should be evident, and there are a normal number of bile ducts. Extramedullary hematopoiesis can be seen.3
Neonatal Acute Liver Failure
The most common etiologies of liver failure present with classic features. Severe coagulopathy and normal aminotransferases are seen in neonatal hemochromatosis. Severe coagulopathy and high aminotransferases are typical of viral hepatitis. Moderate coagulopathy and hepatitis are consistent with metabolic disorders. Metabolic failure, such as hypoglycemia, is common. Presentation in the first weeks of life is an important consideration with neonatal hemochromatosis, mitochondrial hepatopathy, and disseminated herpesvirus infection. Galactosemia and tyrosinemia, in addition to infections, should be considered in the second and third weeks of life. After 3 weeks, disorders to consider include bile acid synthesis defects, vertically transmitted hepatitis B, and hemophagocytic lymphohistiocytosis.4 The underlying mechanism for severe hepatic necrosis is multifactorial and is dependent on patient age, underlying susceptibility, and extent of injury. There should be a high index of suspicion in infants with elevated liver function tests, as well as infants with sepsis or recurrent hypoglycemia. Liver disease can be reversible with acute damage to hepatocytes without destruction of the liver’s capacity to regenerate. In fulminant cases of severe hepatocyte necrosis or in end-stage cirrhotic disease, there are insufficient residual hepatocytes to maintain essential liver function, and the complications are irreversible.
DIFFERENTIAL DIAGNOSIS
Infectious Causes of Hepatitis
Bacteria and Septicemia
Hepatitis in neonatal sepsis typically presents with hepatomegaly and jaundice. Gram-negative bacteria are the most frequent etiologic agents, such as Escherichia coli. Other bacteria include Staphylococcus aureus or Listeria monocytogenes. Typical features include leukocytosis, conjugated hyperbilirubinemia with mild to moderately elevated serum aminotransferase levels, and coagulopathy if disseminated intravascular coagulation is present.
Toxoplasmosis, Rubella, Cytomegalovirus, and Herpes Simplex Virus: The TORCH Infections
Toxoplasmosis: Toxoplasmosis is an intracellular protozoan parasite. Maternal infection occurs by contact with oocytes in cat feces or ingestion of contaminated raw meat. Most newborns are asymptomatic but may present with hepatomegaly, prominent hepatitis, and neurologic findings such as microcephaly, intracranial calcifications, and chorioretinitis.
Rubella: Hepatic involvement is common in congenital rubella, including cholestasis, hepatosplenomegaly, and elevated serum alkaline phosphatase and aminotransferases. Other presenting symptoms include thrombocytopenia, congenital heart disease, cataracts, and neurologic impairments. Hepatic failure is rare. There has been a drastic (over 99%) decline in congenital rubella infections since introduction of the rubella vaccine.5
Cytomegalovirus (CMV): Approximately 10% of CMV-infected newborns have clinical evidence of disease in the neonatal period. More severely infected infants are associated with primary maternal infection in the first trimester. The mortality rate is approximately 20%. Classic symptoms include a petechial rash, hepatosplenomegaly, and jaundice. The hepatitis is typically mild and often resolves within weeks to months, but persistent neurodevelopmental abnormalities cause the most morbidity. CMV is an uncommon cause of ALF, with rare development of cirrhosis and cholestasis leading to liver transplantation. Infection can be treated with ganciclovir and CMV immunoglobulin.6–8
Herpes simplex virus (HSV): Both HSV-1 and HSV-2 cause neonatal infection. HSV is the most common infectious cause of ALF. It can result in severe multisystem infection with encephalopathy. Initial symptoms can be nonspecific, such as lethargy, poor feeding, and fever. Infants often develop a septic-like picture due to disseminated disease with jaundice, hepatomegaly, and ascites. Encephalitis with seizures is present in up to one-third of cases. A vesicular rash can be absent in up to 30% of cases. Laboratory findings include coagulopathy, elevated total and direct bilirubin, and transaminitis. Most neonatal infections are associated with a primary maternal infection late in gestation, but up to 60%–80% of maternal infections are unknown. Liver biopsy reveals necrosis and viral inclusions in intact hepatocytes.9 Mortality is high.
Other congenital infections: Congenital syphilis causing severe infection can present with hepatosplenomegaly and jaundice, and milder cases can have anicteric hepatitis with elevated serum aminotransferases and poor weight gain with snuffles. It is a rare cause of fulminant hepatic failure, and chronic liver disease has not been reported in infants treated appropriately. Tuberculous hepatitis is rare in neonates but can occur through aspiration of infected amniotic fluid or placental spread from miliary tuberculosis. Hepatic lesions show typical caseating necrosis with surrounding giant cells, and the course is often fatal.
Hepatotropic Viruses
Hepatitis A: Hepatitis A is rare in neonates. Incidence in the United States is approximately 25,000 cases per year among all ages. It is a nonenveloped RNA virus, classified as a Picornavirus. The route of transmission is fecal-oral, and the incubation period is 15–50 days. Maternal symptoms and the highest viral titers occur 2 weeks before the illness and up to 1 week after delivery. Most infants and children are asymptomatic (>80%) but can develop fever, malaise, anorexia, nausea, abdominal discomfort, dark urine, and jaundice. Mild elevation in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) and elevated total and direct bilirubin are seen. The clinical illness usually does not last longer than 2 months, although 10%–15% of persons have prolonged or relapsing signs and symptoms for up to 6 months. The virus may be excreted during a relapse. There is no chronic carrier state. Fulminant hepatitis A is rare.10
Hepatitis B: Hepatitis B virus (HBV) is a small, double-shelled virus in the Hepadnaviridae family. Over 90% of neonatal infections occur by vertical transmission through amniotic fluid, vaginal secretions, or maternal blood or postnatally via blood transfusion and prolonged household blood contact. The risk of transmission increases with the presence of hepatitis B e antigen (HBeAg), elevated HBV DNA viral load, acute maternal hepatitis in the third trimester, or a prior infant with chronic infection. As many as 90% of infants who acquire HBV infection via vertical transmission become chronically infected, compared to 30%–50% who become HBV infected when exposed between 1 and 5 years of age. Acute hepatitis B in the neonatal period is rare. The prodromal phase lasts from 3 to 10 days. It is characterized by malaise, anorexia, nausea, vomiting, right upper quadrant abdominal pain, fever, headache, myalgia, skin rashes, arthralgias and arthritis, and dark urine. The icteric phase is variable but usually lasts from 1 to 3 weeks and is characterized by jaundice, light or gray stools, hepatic tenderness, and hepatomegaly (splenomegaly is less common). During convalescence, malaise and fatigue may persist for weeks or months, while jaundice, anorexia, and other symptoms disappear. Fulminant HBV leading to ALF is rare but typically presents at 12 weeks of age. It has been associated with basic core promoter and precore mutants.11
Hepatitis C: Hepatitis C is a small enveloped RNA virus of the family Flaviviridae. Seroprevalence in children in the United States is 0.1%–0.2%. Vertical transmission from mother to fetus is the major route of HCV infection, and the risk is about 5%, with increased risk in maternal human immunodeficiency (HIV) coinfection (3- to 4-fold higher), specific genotypes, and high maternal viral titers. There is no difference in transmission by delivery mode, and transmission through breast milk has not been documented. Most infections are asymptomatic, but acute disease can be associated with jaundice and less transaminitis, as compared to hepatitis B. ALF in infants is rare.12
Hepatitis D: Delta hepatitis virus is a subviral satellite as it can only propagate in the presence of hepatitis B. Transmission is parenteral through blood exposure or illicit drug use. Vertical transmission is unusual. It is not a known cause of ALF.13
Hepatitis E: Hepatitis E is a small nonenveloped RNA virus, and it can cause acute hepatitis in newborns. A study by Khuroo et al revealed that in 15 of 19 babies born to mothers infected with hepatitis E who had positive polymerase chain reaction (PCR) at birth, 37% developed icteric hepatitis, 26% had hepatitis without jaundice, and 26% had high serum bilirubin and normal liver enzymes. Overall, 37% died within the first 7 days of life. The remainder of the infants had self-limited disease without prolonged viremia.14
Other Viral Infectious Causes
Enteroviruses cause mild disease, but immature immune systems create a higher risk for neonates. Multiorgan involvement occurs with encephalomyocarditis (most characteristic of group B coxsackie viruses) and hemorrhage-hepatitis syndrome (most characteristic of echovirus 11).15 Other viruses that can cause neonatal hepatitis include adenovirus, parvovirus B19, human herpesvirus 6 (HHV-6), paramyxovirus, and HIV.
Metabolic and Genetic Causes
Carbohydrate Disorders
Among the carbohydrate disorders, hereditary fructose intolerance is an autosomal recessive disease from defective enzymatic activity of fructose 1,6-bisphosphate aldolase, which is active in the liver, renal cortex, and small intestine. Symptoms such as vomiting, diarrhea, abdominal distension, and failure to thrive develop with introduction of sucrose or fructose. This can progress to hepatomegaly, hemorrhagic disease, and seizures with metabolic acidosis, hyperuricemia, and renal Fanconi syndrome.16
Galactosemia has an incidence of 1:50,000 infants. Features include vomiting, diarrhea, jaundice, poor weight gain, and sometimes sepsis. Hypoglycemia is common. Eye findings can include cataracts and retinal detachment. Despite strict dietary restriction, affected children can have progressive declining cognitive function, speech delay, and other neurologic symptoms. It can lead to acute or chronic liver disease, which often improves after therapy. Most children recover and do well, surviving into adulthood.13
Other carbohydrate disorders, such as fructose 1,6-bisphosphatase deficiency, congenital disorders of glycosylation, glycogen storage disease types IV and IX, and transadolase deficiency, can also cause ALF.17
Amino Acid Disorders
Hereditary tyrosinemia type 1 is an autosomal recessive disorder due to lack of fumaryl acetoacetate hydrolase (FAH), which leads to an accumulation of succinylacetone that damages the liver and kidneys. ALF can present in infancy. Chronic liver disease in combination with rickets and renal Fanconi syndrome occurs with late presentation between 4 and 24 months of age. Significantly elevated α-fetoprotein (AFP) and a classic profile of plasma amino acids are diagnostic. These children are at ongoing risk of developing hepatocellular carcinoma. Other amino acid disorders, such as urea cycle defects, citrin defect, and S-adenosylhomocysteine-hydrolase deficiency can present with ALF.17
Other Genetic Disorders
Deficiency of α1-antitrypsin is the most common genetic cause of neonatal hepatitis. Up to 90% of children with α1-antritrypsin deficiency who develop liver disease present with neonatal hepatitis. Symptoms include severe cholestasis, hepatomegaly, and rarely severe coagulopathy. Approximately half of the infants with liver disease will have progression to cirrhosis. The duration of jaundice is a critical prognostic sign since infants in whom jaundice resolves by 6 months are likely to have a good outcome. A study by Francavilla and coworkers identified that persistence of elevated serum aminotransferases and γ-glutamyl transferase (GGT) through 6–12 months of age and pathologic evidence of bile duct proliferation and bridging fibrosis are markers of rapidly progressive liver disease.18 This is discussed in more detail in the chapter on conjugated hyperbilirubinemia.
There are 3 known types of progressive familial intrahepatic cholestasis (PFIC) due to defects in different transporters in the hepatocyte bile canalicular membrane. All 3 types present with cholestasis and develop diarrhea, pruritis, and fat-soluble vitamin deficiencies. Classic distinguishing features include a normal GGT in PFIC1 and PFIC2 but elevated GGT in PFIC3. Cirrhosis often develops in early childhood, and liver transplantation is required. Please refer to the chapter on conjugated hyperbilirubinemia for more information.13
Trisomy 21 has a known association with transient myeloproliferative disorder. About 20% of patients with transient leukemia develop hepatic fibrosis. A study by Hirabayshi et al identified risk factors of reduced hepatic functional reserve (elevated direct bilirubin and PT and presence of ascites); elevated hyaluronic acid; respiratory failure associated with hepatosplenomegaly; and fibrosis on liver biopsy.19
Neonatal hepatitis is known to occur in trisomy 18 with intrahepatic cholestasis, but there are no clear reports of ALF. A study by Alpert et al examining autopsy cases of 19 infants found an association of neonatal hepatitis with cholestasis and trisomy 18. Of the 3 infants who survived beyond the immediate neonatal period, all had hepatic parenchymal damage with focal necrosis.20
Primary disorders of bile acid synthesis can produce neonatal cholestatic liver disease and progressive neurologic disease. Features include elevated transaminases with normal GGT and liver biopsy with giant cell hepatitis. Neurologic features include spastic paralysis. The most useful screening test is urinary cholanoids (bile acids and bile alcohols). Two inherited defects in the enzymes of bile synthesis include 3β-hydroxysteroid-Δ5-C27-steroid dehydrogenase deficiency and Δ4-3-oxosteroid 5β-reductase deficiency. Early diagnosis is important as these diseases can be treated effectively with supplementation of critical bile acids.21
Additional metabolic and genetic diseases that can be associated with ALF include fatty acid oxidation disorders, Niemann-Pick type C (a disorder of cholesterol esterification), cystic fibrosis, Zellweger syndrome (the reduction or absence of functional peroxisomes), Wolman disease (a deficiency of lysosomal acid lipase), Wilson disease, and neonatal adrenoleukodystrophy.4,22
Mitochondrial Disorders
Mitochondrial disorders should be suspected if there is an association of neuromuscular symptoms with liver dysfunction, multisystem involvement in acute or chronic liver disease, and the presence of lactic acidosis, hepatic steatosis, or ketonemia.23 Mitochondrial hepatopathies involving respiratory chain defects can present as ALF. These can be caused by mutations in the SCO1 gene (required for assembly of complex IV) and mutations in BVS1L (required for assembly of function of complex III). Features include cholestasis, hepatic steatosis, lethargy, hypotonia, vomiting, poor suck, apnea, and seizures. Low hepatic activity of respiratory chain complexes IV (most common), I, III, and occasionally II can be found in these infants. Liver failure progresses to death within weeks to months. Mitochondrial DNA depletion syndromes (MDSs) are other mitochondrial disorders occurring in 2 phenotypes, a myopathic form and a hepatocerebral form. The hepatocerebral form presents in the first weeks of life with vomiting, severe reflux, failure to thrive, developmental delay, and hepatomegaly. The progression of liver failure is less rapid than mitochondrial respiratory chain diseases. Other mitochondrial disorders that can present in infancy include Alpers-Huttenlocher syndrome, Pearson syndrome, and Navajo neurohepatopathy.24
Immunologic Causes
Neonatal hemachromatosis is a unique fetal liver disease and common cause of ALF.25 Intrauterine growth restriction, oligohydramnios, and fetal distress have been documented. Affected infants demonstrate hypoglycemia, extreme cholestasis, severe synthetic dysfunction, and hypoalbuminemia. Serum AST and ALT are typically low, and ferritin and AFP are extremely elevated. Diagnosis rests on abnormally distributed iron deposition in both hepatic and extrahepatic tissues. Abdominal magnetic resonance imaging (MRI) can show abnormal iron signal in the heart, pancreas, spleen, and adrenal glands. The gold standard for diagnosis is to indentify siderosis in extrahepatic tissues, such as submucosal glands, on a buccal mucosal biopsy. Pathology reveals marked fibrosis and cirrhosis, supporting the hypothesis of fetal liver insult. The most likely pathogenic mechanism is a gestational alloimmune disease against a common fetal antigen, evidence exists of increased risk of recurrence in subsequent pregnancies and mothers with affected babies with different fathers.26
Neonatal lupus erythematosus (NLE) is a rare autoimmune disease classically presenting with cardiac disease, specifically congenital heart block, and cutaneous lesions. Maternal autoantibodies, anti-Ro/SSA or anti-La/SSB, are passed transplacentally. A review of a large national registry revealed 9% of infants with NLE had hepatobiliary disease in 3 variants: (1) severe liver failure similar to neonatal hemochromatosis, (2) conjugated hyperbilirubinemia with minimal hepatitis, and (3) mild hepatitis at 2 to 3 months of life. Outside the severe hepatic failure presentation, prognosis was excellent.27 Mild hepatomegaly and splenomegaly can be seen. The hepatic abnormalities can be compounded by congestive heart failure in children with cardiac manifestations of NLE. Elevated liver function tests typically resolve within the first months of life. Pathology tends to resemble that of idiopathic neonatal giant cell hepatitis.28
Oncologic Causes
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening condition due a hyperinflammatory state from high levels of inflammatory cytokines related to impaired function of natural killer cells and cytotoxic T cells.29 Primary familial HLH is inherited in an autosomal recessive pattern, and acquired HLH is thought to be triggered by infection, immune dysregulation, malignancy, or other insult. Clinical criteria include fever, splenomegaly, cytopenia, hypertriglyceridemia, hypofibrinogenemia, and extremely elevated ferritin levels. Additional clinical features include lymphadenopathy, central nervous system (CNS) involvement, and rash. Hepatomegaly and hepatitis often occur early in the disease. Liver pathology is characterized by a variable degree of portal and sinusoidal lymphohistocytic infiltrate with bile duct damage and interface necrosis and inflammation.30 HLH is rapidly progressive and often fatal in the neonatal period without chemotherapy and hematopoietic stem cell transplantation (overall survival 58%). Liver transplantation alone is not curative.31
Neonatal leukemia, neuroblastoma, and hepatic tumors (benign vascular tumors and mesenchymal hamartomas and hepatoblastoma) have also been associated with ALF.13,32,33
Vascular Causes
Shock-liver syndrome is related to reduced hepatic blood flow due to low cardiac output. The typical clinical phases include sudden and transient elevation in serum transaminase levels early in the course of circulatory failure with improvement as the hemodynamic condition is corrected. This is followed by persistent cholestatic jaundice that lasts several weeks. A case report suggested this could be related to the ductus venosus, which may remain patent in the setting of perinatal hypoxemia, leading to a significant reduction in blood flow to the right hepatic lobe, making it more susceptible to ischemia during cardiovascular stress.34
Additional cardiac or vascular abnormalities can lead to ALF, such as myocarditis or development of Budd-Chiari physiology with a portal vein obstruction from thrombotic disorders such as congenital disorders of glycosylation. Other inherited disorders of coagulation to consider in the case of thrombosis include antithrombin III, protein S, and protein C deficiencies and fibrinogen abnormalities.35
Miscellaneous Causes
Various other etiologies are known to cause hepatitis and liver failure in neonates. Medications shown to be hepatotoxic in neonates include valproate, isoniazid, and acetaminophen. Perinatal hypoxia can lead to abnormal liver function, but ALF is rare. Anatomic or structural abnormalities such as extrahepatic bile duct obstruction in biliary atresia and bile duct paucity in Alagille syndrome can cause mild hepatitis and cholestasis but rarely are present with ALF in the neonatal period. Up to 30%–40% of the causes of ALF are indeterminate.
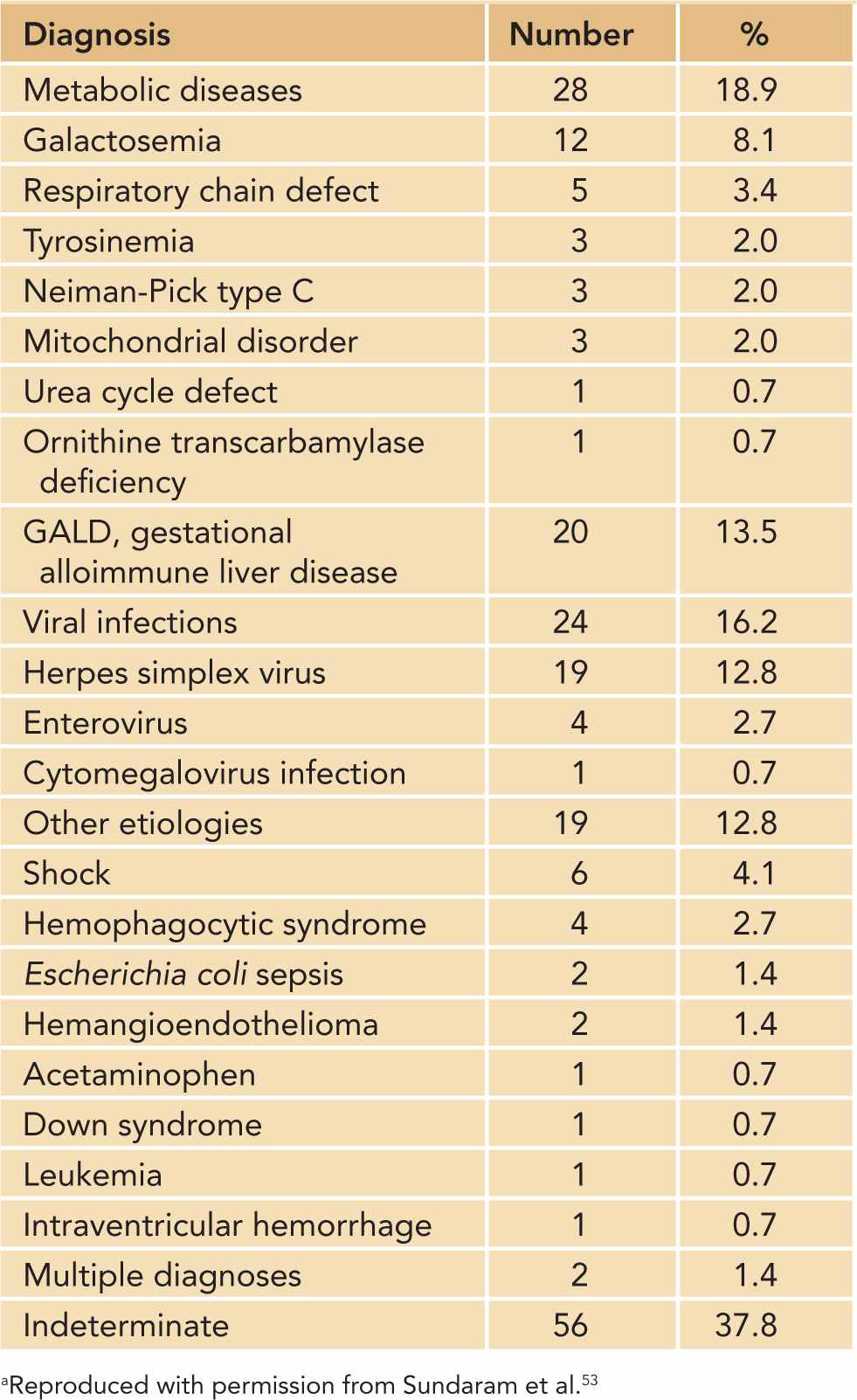
Table 40-1 provides a list of common etiologies of ALF in infants aged 0–90 days.
Table 40-1 Etiologies of Acute Liver Failure in Infants Aged 0–90 Days (n = 148)a
DIAGNOSTIC TESTS
History and Physical Examination
Important history features include consanguinity, onset and symptoms of possible antenatal or perinatal maternal illness, and previous miscarriages or neonatal deaths. Exposure history is important, including family history of tuberculosis, travel history, animal exposures, recent insect bites, and high-risk foods such as unpasteurized dairy food.
A thorough physical examination is critical. Dysmorphic features are important to identify. Irritability and fatigue may be signs of encephalopathy. Scleral icterus may be prominent. A murmur may herald cardiac disease or other genetic syndromes. Other findings, such as pulmonary rales, could indicate pulmonary edema suspicious for cardiac failure. An abdominal examination can reveal hepatomegaly, splenomegaly, or the presence of a fluid wave indicating ascites. An assessment of peripheral edema, wasting, clubbing, or poor peripheral perfusion is helpful. Jaundice, petechiae, ecchymosis, or spider angiomas are important to identify on a skin examination.
Laboratory Tests
First Tier
Laboratory: An initial workup should include basic hematology and chemistry tests, such as complete blood cell count (CBC) with differential, blood type, and Coombs; complete metabolic panel, including glucose, liver function tests such as total and direct bilirubin, AST, ALT, alkaline phosphatase, and GGT. Liver synthetic function should be evaluated in all infants with a coagulation profile, fibrinogen, factor levels (V, VII, and VIII), and albumin. Iron studies and ferritin should be also sent.
Microbiology: The key is to focus on direct identification of possible infectious agents through specific immunoglobulin (Ig) M antibodies, viral titers, PCR-based diagnostics, or culture. Key screening should include hepatotropic viruses with anti-HAV (anti-hepatitis A virus) IgM, hepatitis B surface antigen (HBsAg) and anti-HBs, and hepatitis B core antibody (anti-HBc) IgM. Maternal testing involves HBsAg, HBeAg, hepatitis B e antibody (anti-HBe), anti-HBc, anti-hepatitis C IgM, and hepatitis C PCR. Evaluation for HSV includes HSV PCR on cerebrospinal fluid (CSF) or culture of skin lesions plus eye, oropharyngeal, and rectal cultures. HIV enzyme-linked immunosorbent assay (ELISA) should be sent. CMV and Epstein-Barr virus (EBV) serology or quantitative PCR should also be considered. Other viruses may be identified by respiratory direct fluorescence assay (DFA).
Urine studies: Urine culture and urine CMV antigen test should be sent. An initial metabolic workup should include urine reducing substances (positive in galactosemia), urine succinylacetone (elevated in tyrosinemia), and urine organic acids. Urine toxicology is important to identify potential exposures.
Second Tier (Based on Suspected Diagnosis)
Metabolic and genetic: A basic metabolic workup may include assessment of lactate, pyruvate, ammonia, and a venous blood gas. Plasma acylcarnitine profile, serum amino acids, and α1-antritypsin serum level and phenotype are also commonly sent. The initial screen for galactosemia is urine reducing substances, which are nonspecific and can be positive in prematurity or negative in infants too sick for oral feedings. Confirmation of galactosemia is made by a quantitative assay of erythrocyte galactose-1-phosphate uridyl transferase (GALT) enzyme activity. This must be sent prior to blood transfusion. Diagnosis can also be confirmed by aldolase activity in a liver biopsy specimen. A traditional fructose tolerance test is contraindicated because of the risk of hypoglycemia. Hereditary tyrosinemia is often associated with an elevated AFP, between 40,000 and 70,000 µg/L. The plasma amino acid profile will show elevations in tyrosine, phenyalanine, and methionine. There is positive succinylacetone in the urine. Genetic diagnoses may be identified by karyotype or chromosomal genomic hybridization. Molecular resequencing arrays can be sent for known mutations in Alagille syndrome, PFIC, and α1-antitrypsin deficiency. Urine quantitative bile acids determination by mass spectrometry can screen for disorders of bile synthesis.
Immunologic: Neonatal hemochromatosis is characterized by high serum ferritin and high serum AFP and confirmed by demonstrating extrahepatic iron deposits sparing the reticuloendothelial system.
Oncologic: The diagnosis of hemophagocytic lymphohistiocytosis can be made by fulfillment of 5 of 8 criteria: fever, splenomegaly, cytopenias, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis in the bone marrow, abnormal natural killer cell functional assay, elevated soluble interleukin (IL) 2R α level (>2400 U/mL), and elevated ferritin level (>50 µg/L). Molecular analysis for known mutations in familial HLH could also be performed.36
Mitochondrial: Mitochondrial disorders can be identified by a markedly elevated plasma lactate, an elevated molar ratio of plasma lactate to pyruvate (>20), and an elevated β-hydroxybutyrate. A distinguishing feature of MDS is a low ratio (<10%) of the normal amount of mitochondrial DNA (mtDNA) relative to nuclear DNA in the affected tissues and a normal mtDNA sequence.24
Table 40-2 provides additional details on laboratory testing.
Table 40-2 General Investigation of Neonatal Liver Failurea



