Congenital Adrenal Hyperplasia
DEFINITION/CLASSIFICATION
Congenital adrenal hyperplasia (CAH) is a family of inherited, autosomal recessive disorders of adrenal steroidogenesis due to an abnormality in a step necessary for conversion of cholesterol to cortisol in the adrenal cortex (Figure 46-1). Of all CAH cases, 95% are caused by 21-hydroxylase deficiency. There are 2 forms of 21-hydroxylase deficiency: classic and nonclassic CAH (also called late-onset CAH). Classic CAH comprises the salt-wasting form (cortisol deficiency and aldosterone deficiency) and simple virilizing form. The salt-wasting form comprises 75% and simple virilizing form 25% of patients with 21-hydroxylase deficiency. The classic form is associated with severe enzyme deficiency, leading to prenatal virilization in girls; the nonclassic form has mild enzyme deficiency that causes postnatal hyperandrogenism but no prenatal virilization.
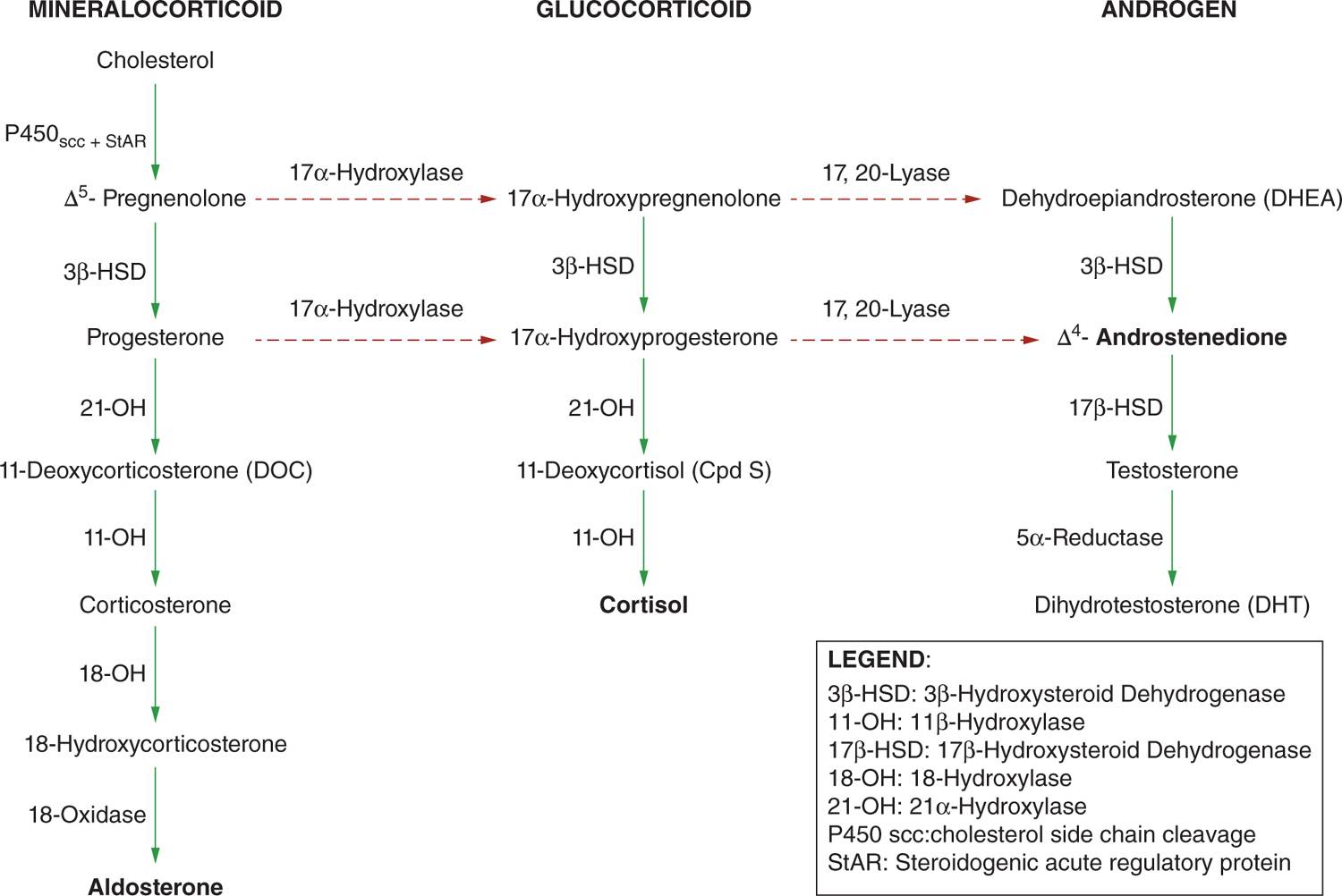
FIGURE 46-1 Adrenal biosynthesis pathway.
ETIOLOGY
The most common form of CAH is caused by mutations in CYP21A2, the gene encoding the adrenal steroid 21-hydroxylase enzyme (P450c21).1 The steroid 21-hydroxylase enzyme catalyzes conversion of 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol and progesterone to deoxycorticosterone, which are precursors of cortisol and aldosterone, respectively. In 21-hydroxylase deficiency, because of deficient cortisol synthesis, progesterone and 17-hydroxyprogesterone are shunted to the androgen synthetic pathway (androstenedione and testosterone). These adrenal androgens are oversecreted in utero, resulting in variable degrees of virilization in the female fetus.
EPIDEMIOLOGY
The incidence of classic CAH in the general population, as shown by newborn screening in different populations worldwide, ranges from 1:10,000 to 1:20,000, with an overall incidence of 1:15,000.2,3 The incidence of CAH in specific populations ranges from 1 in 5000 live births in Saudi Arabia to 1 in 21,270 live births in New Zealand. The newborn screening incidence in the United States and Canada is 1 in 14,203 live births, with Brazil at 1 in 1863 live births and Japan at 1 in 18,827. A high frequency of CAH exists among Yupik Eskimos from western Alaska: 1 in 282 live births.
The prevalence of nonclassic 21-hydroxylase deficiency CAH in the general heterogeneous population of New York City was estimated to be 1 in 100. Ashkenazi Jews have the highest prevalence at 1/27. Other ethnic groups with a high prevalence of nonclassic CAH include Hispanics (1/40), Slavs (1/50), and Italians (1/300).1
CLINICAL MANIFESTATIONS
The cardinal feature of classic CAH in newborn females is ambiguous genitalia (Figure 46-2). Clitoromegaly occurs as a result of adrenal androgens binding to genital skin androgen receptors. Females may present with a urogenital sinus (Figure 46-3). Urogenital sinus due to high levels of circulating adrenal androgens beginning at about 7 weeks’ gestation prevent the formation of separate vaginal and urethral canals. Females with classic CAH have normal Müllerian structures (uterus, fallopian tubes). The girl with classic CAH has ambiguous or male-appearing external genitalia with perineal hypospadia, chordee, and cryptorchidism. The severity of virilization is graded using Prader staging, a 5-point scale developed by Prader, with stage 1 indicating mild clitoromegaly and stage 5 representing a cryptorchid male with penile urethra.
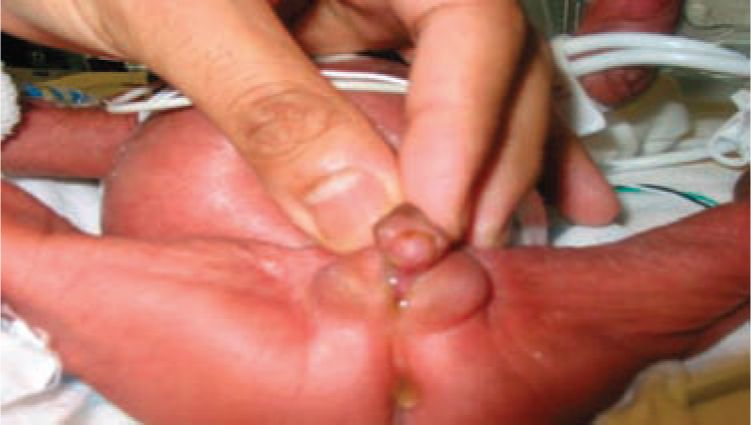
FIGURE 46-2 Ambiguous genitalia in a girl with classic congenital adrenal hyperplasia (CAH): scrotalization/pigmentation of the labia majora, labial fusion, and clitoromegaly.
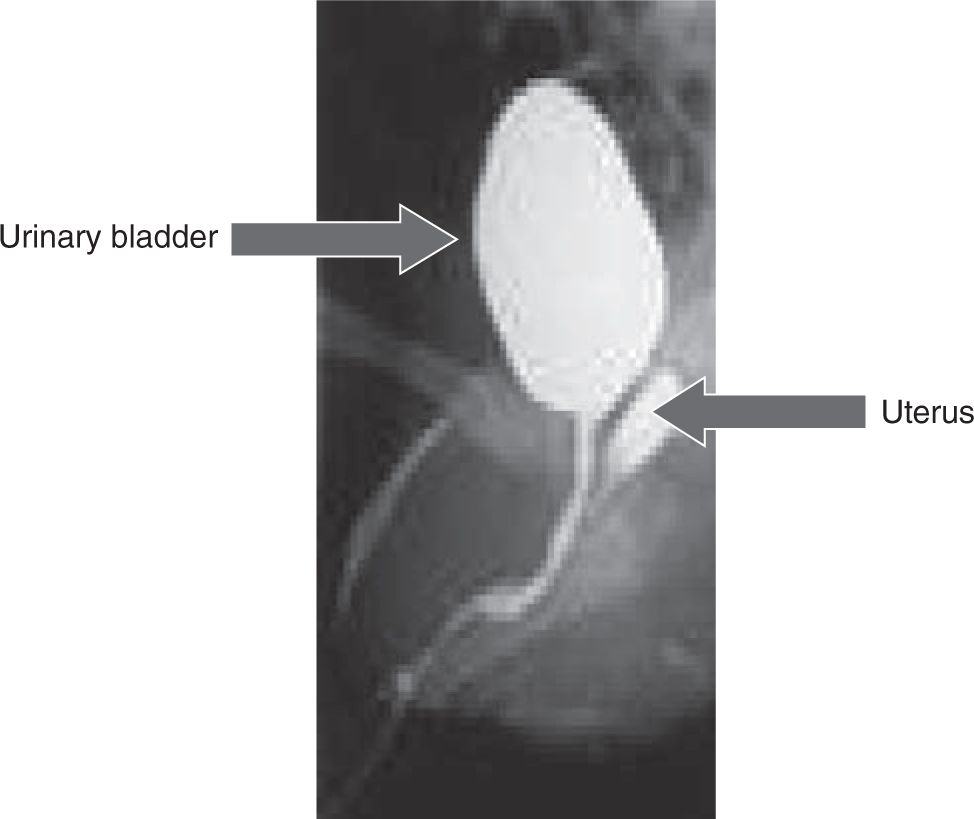
FIGURE 46-3 Genitogram showing the uterus and bladder in a girl with classical congenital adrenal hyperplasia (CAH).
Males with classic CAH do not have ambiguous genitalia but may present with subtle scrotal hyperpigmentation because of corticotropin (ACTH) hypersecretion or phallic enlargement because of excessive adrenal androgens in utero.
Neonates with classic salt-wasting CAH typically manifest salt-wasting crisis during the first 1–3 weeks of life. Salt-wasting crisis may occur as late as a few months of age. This manifests as poor feeding, vomiting, weak cry, failure to thrive, loose stools, dehydration, and lethargy. The biochemical sine qua non of salt-wasting crisis is hyponatremia, hyperkalemia, and metabolic acidosis with or without hypoglycemia. Without treatment, circulatory collapse, arrhythmias from hyperkalemia, and death may occur within days or weeks.
Postnatally in untreated boys and girls, excessive adrenal androgen secretion leads to rapid somatic growth, bone age advancement, progressive penile or clitoral enlargement, and premature appearance of pubic hair, axillary or facial hair, and acne. Without cortisol treatment, early epiphyseal closure occurs as a result of aromatization of adrenal androgens (androstenedione and testosterone to estrone and estradiol, respectively) and results in short adult height.
DIAGNOSIS
CAH is the most common cause of ambiguous genitalia in the newborn, especially if no gonads are palpable in the scrotum or inguinal canal. Timely diagnosis is needed to prevent sex misassignment of a female newborn as male. If gonads are palpable in the scrotum or inguinal canal, workup for 46 XY disorder of sex differentiation or other causes of ambiguous genitalia should be pursued.
Immediate steroid treatment is necessary to prevent salt-wasting crisis in the salt-wasting form of CAH. Karyotype will identify chromosomal sex, and pelvic ultrasonography will help identify Müllerian structures (ovaries and uterus). Consultation with an endocrinologist should be sought. Adrenal steroids should be measured by blood sample on day 2–3 of life. A markedly elevated level of 17-OHP (>10,000 ng/dL or > 300 nmol/L) in a virilized 46, XX female with nonpalpable gonads is most likely due to CAH.
Diagnosis of 21-hydroxylase deficiency is most accurately established by measuring 17-OHP before and 30 or 60 minutes after intravenous injection of 0.25 mg of Cosyntropin (ACTH1-24). Basal and 60-minute 17-OHP are plotted on a nomogram and distinguish normal individuals and patients with nonclassical and classical CAH.
In affected females, androstenedione and testosterone levels are high. In males, testosterone is usually elevated because of minipuberty in the first 6 months of life and is thus not helpful. ACTH levels are high but are not needed for diagnosis. In salt-wasting CAH, plasma renin is elevated and serum aldosterone is inappropriately low for the renin level.
TREATMENT
Cortisol deficiency is treated with glucocorticoids, usually in the form of hydrocortisone. Glucocorticoid treatment also suppresses high adrenal androgens, preventing further virilization, rapid somatic growth, and early epiphyseal fusion.4
Hydrocortisone tablets at a dose of 10–15 mg/m2/d in 2 or 3 divided doses are the preferred treatment. Infants with CAH require higher doses, 50 mg/m2/d divided in 2 to 4 doses during the first few weeks of life to suppress ACTH hypersecretion. Patients with salt-wasting CAH require mineralocorticoid replacement with fludrocortisone 0.1–0.3 mg daily in 1 or 2 divided doses and NaCl supplementation at 1–3 g/d. NaCl tablets are available, and a 1-g tablet is equivalent to 17 mEq of NaCl. Table salt (one-quarter teaspoon = 575 mg NaCl) can also be added to formula if NaCl tablets are not available.
Stress dosing (3–5 times the maintenance dose) is needed in febrile illness (>38.5°C), gastroenteritis with dehydration, surgery accompanied by general anesthesia, and major trauma. Parenteral Solu-Cortef® (hydrocortisone sodium succinate) 25 mg IM or IV should be given for severe vomiting, dehydration, surgery, or severe trauma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


