Chronic Anovulation and the Polycystic Ovary Syndrome
 |
Anovulation is very common and has a number of different clinical manfestations, including amenorrhea, dysfunctional uterine bleeding, and hirsutism. The condition also has serious potential consequences, such as infertility and an increased risk for developing endometrial hyperplasia and neoplasia. In many anovulatory women, the pathophysiology involves insulin resistance, which increases the risks for developing diabetes mellitus and cardiovascular disease. In others, overt hypogonadism increases the risk for developing early osteoporosis. All clinicians who care for women must therefore be thoroughly familiar with the evaluation and management of anovulatory women.
Normal ovulatory function requires coordination at all levels of the hypothalamic-pituitarygonadal axis, and anovulation can result from disruption at any level. This chapter considers the variety of mechanisms that can cause anovulation and the clinical consequences of chronic anovulation, focusing on the most common anovulatory disorder, the polycystic ovary syndrome, and its management.
Causes of Anovulation
The complex interaction of neuroendocrine, intraovarian, and endometrial mechanisms that regulate the normal ovulatory menstrual cycle are discussed in detail in other chapters in this text (Chapters 5 and 6). They are summarized briefly here, to provide the foundation for subsequent discussion of the pathophysiology of anovulation.
As the corpus luteum regresses and the menstrual cycle draws to a close in the late luteal phase, serum concentrations of estradiol, progesterone, and inhibin A decline to basal levels, releasing the hypothalamic-pituitary axis from their collective negative
feedback effects. Accordingly, the frequency of hypothalamic gonadotropin-releasing hormone (GnRH) secretion increases, stimulating an increase in pituitary folliclestimulating hormone (FSH) secretion, which serves to “recruit” a new cohort of small antral follicles or, more accurately, to rescue a group of follicles from otherwise programmed demise via apoptosis. During the early follicular phase, the serum concentration of inhibin B, secreted by the recruited pool of small antral follicles, rises progressively.
feedback effects. Accordingly, the frequency of hypothalamic gonadotropin-releasing hormone (GnRH) secretion increases, stimulating an increase in pituitary folliclestimulating hormone (FSH) secretion, which serves to “recruit” a new cohort of small antral follicles or, more accurately, to rescue a group of follicles from otherwise programmed demise via apoptosis. During the early follicular phase, the serum concentration of inhibin B, secreted by the recruited pool of small antral follicles, rises progressively.
During the midfollicular phase, ovarian autocrine and paracrine mechanisms involving activin and insulin-like growth factors enhance FSH-stimulated aromatase activity in granulosa cells to help create and sustain the estrogenic microenvironment required for continued follicular growth and development. Whereas an estrogenic follicular milieu fosters further growth, an androgenic milieu promotes atresia. As the serum inhibin B concentration reaches its peak, estradiol and inhibin A levels, derived from the granulosa cells of growing follicles in the cohort, begin increasing steadily. In response to their combined inhibitory effects, luteinizing hormone (LH) pulse amplitude decreases and pulse frequency increases (presumably reflecting the pattern of hypothalamic GnRH secretion), and serum concentrations of both FSH and LH fall gradually. Declining FSH levels remain sufficient to support continued growth of the selected dominant follicle, which has more granulosa cells and FSH receptors and a more advanced microvasculature, but become inadequate to support further development in smaller follicles in the cohort.
During the late follicular phase, inhibin A and insulin-like growth factors combine to promote LH-stimulated androgen production in theca cells, which provides substrate for aromatization to estrogen in the proliferating mass of granulosa cells within the preovulatory follicle. FSH and estradiol then combine to induce expression of LH receptors on the granulosa cells that will mediate luteinization and ovulation when the follicle reaches full maturity. UItimately, serum levels of estradiol derived from the preovulatory follicle exceed the threshold concentration required to exert positive feedback effects centrally, acting primarily on the pituitary to induce the midcycle LH surge. The LH surge completes follicular maturation and triggers a cascade of events resulting in extrusion of the oocyte and formation of the corpus luteum. The oocyte completes the first meiotic division and local secretion of plasminogen activator and other cytokines mediates erosion of the follicular wall, allowing the oocyte to emerge with its surrounding investment of cumulus cells. The mural granulosa cells begin to luteinize and produce progesterone.
After ovulation, serum estradiol concentrations fall precipitously, but only transiently, before rising again in parallel with progesterone and inhibin A produced by the corpus luteum. Progesterone transforms the endometrium from a proliferative to a secretory morphology and stimulates a still uncharacterized cascade of biochemical events that renders the endometrium receptive to embryo implantation. As the progesterone level rises to its peak during the midluteal phase, LH pulse frequency decreases again and gonadotropin levels fall progressively to their nadir in the late luteal phase. Unless pregnancy intervenes and rapidly rising levels of human chorionic gonadotropin (hCG) rescue the corpus luteum and stimulate continued high level progesterone secretion, the corpus luteum regresses, estradiol and progesterone levels fall, support for the endometrium is withdrawn, and menses ensue.
Central Defects
Although difficult to demonstrate, hypothalamic dysfunction offers both a logical and likely explanation for ovulatory failure. A normal pituitary response to feedback signals from the follicle requires pulsatile GnRH secretion within a critical range. The onset of
puberty in girls results from decreasing central inhibition of GnRH neuronal activity and increasing pulsatile GnRH secretion, which stimulates a progressive increase in pituitary gonadotropin release and, in turn, ovarian follicular growth and estrogen production (Chapter 10). After menarche, cycle length and menstrual characteristics in adolescent girls typically vary until the hypothalamic-pituitary-ovarian axis matures and the positive feedback relationship between estradiol and gonadotropin secretion and ovulation becomes established. Factors that reactivate central inhibitory mechanisms, such as emotional, nutritional (weight loss, eating disorders), or physical stress (excessive exercise), can suppress GnRH neuronal activity, leading to dysfunctional patterns of gonadotropin secretion that fail to promote progressive follicular development, resulting in anovulation. Although such patients more commonly present with amenorrhea (Chapter 11), lesser degrees of GnRH neuronal suppression can result in homeostatic levels of pituitary-ovarian function and a euestrogenic chronic anovulatory state.
puberty in girls results from decreasing central inhibition of GnRH neuronal activity and increasing pulsatile GnRH secretion, which stimulates a progressive increase in pituitary gonadotropin release and, in turn, ovarian follicular growth and estrogen production (Chapter 10). After menarche, cycle length and menstrual characteristics in adolescent girls typically vary until the hypothalamic-pituitary-ovarian axis matures and the positive feedback relationship between estradiol and gonadotropin secretion and ovulation becomes established. Factors that reactivate central inhibitory mechanisms, such as emotional, nutritional (weight loss, eating disorders), or physical stress (excessive exercise), can suppress GnRH neuronal activity, leading to dysfunctional patterns of gonadotropin secretion that fail to promote progressive follicular development, resulting in anovulation. Although such patients more commonly present with amenorrhea (Chapter 11), lesser degrees of GnRH neuronal suppression can result in homeostatic levels of pituitary-ovarian function and a euestrogenic chronic anovulatory state.
Pituitary Tumors
Pituitary tumors can cause anovulation by inhibiting gonadotropin secretion. They may compress pituitary gonadotrophs directly, or interrupt delivery of hypothalamic GnRH by compression of the pituitary stalk. Alternatively, they may cause hyperprolactinemia by interfering with the inhibitory actions of hypothalamic dopamine (the putative prolactin inhibitory hormone) on pituitary lactotrophs, resulting in a secondary suppression of pulsatile GnRH secretion.
Hyperprolactinemia
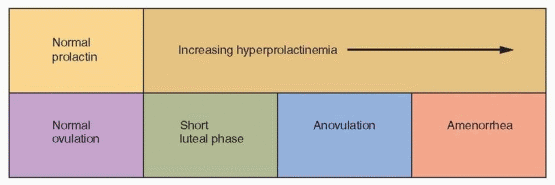
Hyperprolactinemia is another specific example of anovulation resulting from a central defect.1 The mechanism involves disruption or inhibition of the normal GnRH pulse rhythm, resulting in ineffective or frankly low levels of gonadotropin secretion. It’s possible that elevated prolactin levels stimulate a generalized increase in hypothalamic dopaminergic activity, intended to suppress prolactin secretion but also inhibiting GnRH neurons. In any event, increasing prolactin levels can result in a spectrum of ovulatory dysfunction, ranging from a short luteal phase to anovulatory cycles to amenorrhea and hypogonadotropic hypogonadism, depending on the extent to which gonadotropin secretion is disturbed or suppressed. Mild hyperprolactinemia may cause only a short luteal phase, resulting from inadequate preovulatory follicular development.2,3 Moderate hyperprolactinemia frequently causes oligomenorrhea or amenorrhea, and higher prolactin levels typically result in frank hypogonadism with low estrogen levels.4,5 A breast examination with gentle compression looking for evidence of galactorrhea and measurement of the serum prolactin concentration are important parts of the evaluation of all anovulatory women.
 |
Abnormal Gonadotropin Secretory Dynamics
Many, but not all anovulatory women with polycystic ovaries exhibit abnormal gonadotropin secretory dynamics. The most common abnormality is an increase in mean serum LH levels, due to an increase in both LH pulse frequency and amplitude.6,7 Serum concentrations of FSH typically are normal or low. The pattern could result from a decrease in hypothalamic dopamine or opioid inhibition of pulsatile GnRH secretion,8 or from abnormalities in steroid hormone feedback, including the lack of progesterone (due to anovulation)9 or increased circulating androgen levels.10 Other evidence from studies in nonhuman primates and women suggests strongly that prenatal exposure to increased androgen concentrations induced by genetic and/or environmental factors may program the GnRH pulse generator in the female fetus in such a way as to result in increased pituitary LH secretion, causing disordered follicular development and ovarian hyperandrogenism.11,12 and 13 The increased prevalence of chronic anovulation and polycystic ovaries in women with epilepsy offers another example of how central nervous system dysfunction can disrupt the hypothalamicpituitary-ovarian axis and result in anovulation.14,15
Abnormal Feedback Signals
Anovulation can result from abnormal estrogen feedback signals from the periphery, in two ways. Chronically elevated estrogen levels may not permit the increase in FSH secretion required to stimulate or sustain progressive follicular development. Conversely, poor follicular development may not generate or sustain the estradiol level required to induce the ovulatory LH surge.
Chronically Elevated Estrogen Concentrations
The fall in estradiol levels that normally occurs during the late luteal phase (as the corpus luteum regresses) is a prerequisite for the inter-cycle rise in FSH that drives the wave of new follicular development. Sustained high levels of estrogen negative feedback caused by increased production or decreased clearance and metabolism can prevent any significant increase in FSH levels, resulting in a chronic anovulatory state.
Pregnancy is the most common and obvious example of anovulation resulting from sustained high levels of estrogen production. Rare estrogen producing ovarian tumors (e.g., granulosa cell tumors) can have the same effect. Although the adrenals do not normally secrete appreciable amounts of estrogen directly into the circulation, they contribute via their secretion of androgens (androstenedione, dehydroepiandrosterone and its sulfate), which can be converted to estrogen in the periphery. Adipose tissue has significant aromatase activity, which converts androgens to estrogens,16 thereby providing at least one mechanism for the well-known association between obesity and chronic anovulation (see below).
The clearance and metabolism of estrogen can be impaired in a variety of conditions, such as thyroid or hepatic disease. Both hyperthyroidism and hypothyroidism can cause chronic anovulation by altering the metabolic clearance and peripheral interconversion of steroid hormones.17,18 and 19 Hypothyroidism can be associated with elevated prolactin levels, providing the rationale for measuring serum thyroid-stimulating hormone (TSH), as well as prolactin, in the evaluation of anovulatory and amenorrheic women. Hepatic disease also disturbs the normal clearance and metabolism of sex steroids.20
Failure of the LH Surge
The rising tide of estradiol arising from the preovulatory follicle in the late follicular phase induces the midcycle LH surge that stimulates ovulation. Quite obviously, women with gonadal dysgenesis or ovarian failure are anovulatory because they have no remaining functional ovarian follicles and no significant estrogen production. More commonly, clinicians encounter patients with normal serum levels of gonadotropins and estradiol who do not ovulate, whose anovulation results from the failure to achieve complete follicular development and to generate and sustain the level of estradiol required to induce the LH surge. Normal women typically also become anovulatory during the years immediately preceding the menopause, probably reflecting intrinsic deficiencies in aging follicles that impair normal follicular maturation.
Local Ovarian Conditions
A disturbance in one or more of the delicately balanced intra-ovarian regulatory mechanisms that serve to select the dominant follicle and allow it alone to grow and develop in the face of declining levels of FSH may lead to anovulation. Activins, inhibins, and insulin-like growth factors act via local autocrine and paracrine mechanisms to first enhance the action of FSH by increasing the concentration of FSH receptors within the dominant follicle, then combine to induce the appearance of LH receptors required to mediate the actions of LH during the midcycle surge that drives the final stages of follicular maturation and stimulates ovulation. A follicle can thus fail to grow and ovulate due to a failure or interference with any of these local mechanisms (Chapter 6).
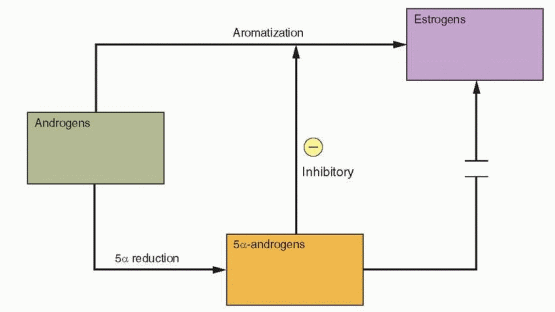
The “two-cell, two gonadotropin” concept of ovarian follicular development (Chapters 2 and 6) emphasizes the critical importance of local androgen concentrations. At low levels, androgens serve as substrate for FSH-induced aromatization and estrogen production. At higher concentrations, androgens are converted alternatively to more potent 5α-reduced androgens, which cannot be converted to estrogen and also inhibit aromatase activity and FSH induction of LH receptors on granulosa cells. Consequently, abnormally high local androgen concentrations, from any cause, impede follicular maturation, promote atresia, and predispose to a chronic anovulatory state.
 |
Obesity
The prevalence of obesity in women with chronic anovulation and polycystic ovaries is high, ranging between 35% and 60%.21,22,23 and 24 Obesity predisposes to chronic anovulation in at least three distinct ways:
Increased peripheral aromatization of androgens, resulting in chronically elevated estrogen concentrations.
Decreased levels of hepatic SHBG production, resulting in increased circulating concentrations of free estradiol and testosterone.
Insulin resistance, leading to a compensatory increase in insulin levels that stimulates androgen production in the ovarian stroma, resulting in high local androgen concentrations that impair follicular development.
Defining the Cause of Anovulation
Whereas the cause of anovulation may be relatively clear in women with ovarian failure, pituitary tumors, eating disorders, hyperprolactinemia, or obesity, frequently it is not possible to isolate the specific mechanism responsible. However, it also is often not necessary. Regardless of its cause, the clinical manifestations and consequences are predictable, easily documented, and generally not difficult to manage. Women with absent or abnormal menstrual function who are otherwise healthy can be categorized as follows:
Ovarian failure. Hypergonadotropic hypogonadism, reflecting the inability of the ovary to respond to gonadotropin stimulation, due to follicular depletion (Chapter 11).
Central defects. Hypogonadotropic hypogonadism, reflecting hypothalamic or pituitary failure or suppression (Chapter 11).
Hypothalamic-pituitary-ovarian dysfunction, resulting in asynchronous gonadotropin and estrogen production, having a wide variety of causes and clinical manifestations that depend on the level of ovarian function, including amenorrhea (Chapter 11), hirsutism (Chapter 13), dysfunctional uterine bleeding (Chapter 15), endometrial hyperplasia and cancer (Chapter 18), and infertility (Chapters 27 and 31).
The polycystic ovary syndrome (PCOS) is the most obvious and common condition associated with chronic anovulation, affecting 4-6% of reproductive age women.29,30 Several mechanisms contribute to the pathophysiology of anovulation in PCOS, operating at every level of the reproductive system. It is inaccurate to state that PCOS is the most common “cause” of anovulation, because PCOS does not cause anovulation; rather, PCOS is the consequence of chronic anovulation, which can result from a wide variety of causes. In that context, the disorder is described more accurately as chronic anovulation with
polycystic ovaries. Although the term PCOS is now firmly established in our scientific and clinical lexicon, it is important to emphasize that PCOS is not a discrete or specific endocrine disorder having a unique cause or pathophysiology. Instead, the condition is best viewed as a final common pathway in the chronic anovulatory state.
polycystic ovaries. Although the term PCOS is now firmly established in our scientific and clinical lexicon, it is important to emphasize that PCOS is not a discrete or specific endocrine disorder having a unique cause or pathophysiology. Instead, the condition is best viewed as a final common pathway in the chronic anovulatory state.
The Polycystic Ovary Syndrome
Multicystic or “sclerocystic” ovaries were recognized as early as the mid-18th century, but associated primarily with pelvic pain or menorrhagia. In the early 20th century, prevailing hypotheses viewed them as resulting from inflammation due to infection, congestion due to pressure or partial torsion that disrupted normal blood flow to the ovary, or from dystrophy due to abnormalities in ovarian nutrition.31
In 1935, Irving F. Stein and Michael L. Leventhal first described a symptom complex associated with anovulation.32 Both gynecologists were born in Chicago, both were graduates of Rush Medical College, and both spent their entire professional careers at the Michael Reese Hospital.33 Stein and Leventhal described seven patients (four being obese) with amenorrhea, hirsutism, and enlarged, polycystic ovaries. They reported that all seven resumed regular menses and that two became pregnant after bilateral ovarian wedge resection, involving the removal of one-half to three-fourths of each ovary. Stein and Leventhal developed the wedge resection procedure after observing a resumption of menses following ovarian biopsy in several patients with amenorrhea. They speculated that the thickened ovarian capsule prevented follicles from reaching and escaping from the surface of the ovary.
Careful histologic studies of the “Stein-Leventhal ovary” revealed that they had twice the cross-sectional area of normal ovaries, the same number of primordial follicles, double the number of developing and atretic follicles, a 50% thicker and more collagenized tunica, a 5-fold thicker subcortical stroma, and a 4-fold greater number of hilar cell “nests” than normal ovaries. These studies further suggested that “hyperthecosis,” characterized by an abundance of such nests and a markedly increased stroma, was likely just a later or more advanced stage of a progressive process.34
The pathophysiology responsible for development of polycystic ovaries has puzzled gynecologists and endocrinologists for many years and proven very difficult to define. However, there is an answer that is very simple, logical, and clinically useful. The characteristic polycystic ovary develops when a chronic anovulatory state persists for a sufficient length of time. A cross section of anovulatory women at any one point in time will demonstrate that approximately 75% have multicystic or polycystic ovaries.24,35 Because there are many causes of anovulation, there are many causes of polycystic ovaries. Any of the causes of anovulation outlined earlier can yield the same or a similar clinical presentation. The polycystic ovary results from a functional derangement, not from a specific central or local defect.
Pathophysiology
Whereas the morphological characteristics of polycystic ovaries were attributed at first to pathological changes in the ovaries themselves that prevented ovulation,34 they now are recognized as reflecting the disordered endocrine milieu that results from chronic
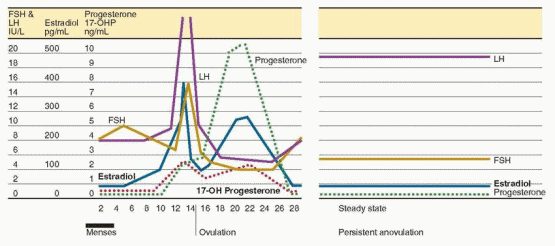
anovulation. In contrast to the cyclic pattern of hormone concentrations that occurs during the normal cycle, the endocrine milieu in women with chronic anovulation is characterized by a “steady state” in which gonadotropin and sex steroid concentrations vary relatively little, by comparison.
anovulation. In contrast to the cyclic pattern of hormone concentrations that occurs during the normal cycle, the endocrine milieu in women with chronic anovulation is characterized by a “steady state” in which gonadotropin and sex steroid concentrations vary relatively little, by comparison.
The average daily production of both androgens and estrogens is increased in women with PCOS, as reflected by elevated serum concentrations of testosterone, androstenedione, dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), 17α-hydroxyprogesterone (17-OHP), and estrone. The results of treatment with a long-acting GnRH agonist (aimed at suppressing gonadotropin-dependent ovarian steroid production) indicate that the increases in serum testosterone, androstenedione, and 17-OHP derive from the ovary and are LH-dependent, whereas those in DHEA and DHEA-S derive from the adrenal.36,37,38 and 39 Serum estrone concentrations are modestly elevated, due to peripheral conversion of increased amounts of androstenedione. In contrast, serum estradiol levels in women with PCOS fluctuate but generally remain within the range typically observed in the early follicular phase,40 reflecting continued low-level production from limited follicular development.41,42
The endocrine milieu in women with PCOS reflects the chronic anovulatory state, which may result from a wide variety of causes. Current perspectives view PCOS as a complex disorder, similar to cardiovascular disease and type 2 diabetes mellitus, wherein numerous genetic variants and environmental factors interact, combine, and contribute to the pathophysiology.43 Not surprisingly, attention has focused on identifying genetic variants involving the regulation of gonadotropin secretion and action, insulin secretion and action, weight and energy regulation, and androgen synthesis and action.
 |
Gonadotropin Secretion and Action
Stein and Leventhal suggested that polycystic ovaries were likely to result from abnormal anterior pituitary hormonal stimulation,32 based on earlier observations that treatment with a urinary extract of anterior pituitary hormones could induce changes similar to those in polycystic ovaries.44 Subsequent studies employing an LH bioassay (based on the ovarian
response of immature female rats or the prostatic response of hypophesectomized male rats to urinary extracts) demonstrated excessive LH activity in women with PCOS,45,46,47 and 48 which later was confirmed by studies using a radioimmunoassay.49
response of immature female rats or the prostatic response of hypophesectomized male rats to urinary extracts) demonstrated excessive LH activity in women with PCOS,45,46,47 and 48 which later was confirmed by studies using a radioimmunoassay.49
Compared to normally cycling women, those with PCOS generally exhibit increased serum LH concentrations, low-normal FSH levels, and increased LH:FSH ratios.7,50,51 The increase in serum LH levels results from abnormal LH secretory dynamics, characterized by an increase in LH pulse frequency, and to a lesser extent, also in pulse amplitude.6,52,53 and 54 The decrease in FSH levels results from the increase in GnRH pulse frequency, the negative feedback effects of chronically elevated estrone concentrations (derived from peripheral aromatization of increased androstenedione), and normal or modestly increased levels of inhibin B (derived from small follicles).55,56
LH pulse frequency in women with PCOS does not exhibit the normal cyclic variation seen in ovulatory women and is relatively constant, at approximately one pulse per hour. The pattern presumably reflects a similar increase in hypothalamic GnRH pulse frequency, which favors secretion of LH more than FSH.57,58 and 59 The LH response to an acute exogenous GnRH stimulus also is exaggerated in women with PCOS, but to a lesser extent in obese than in lean women; accordingly, LH pulse amplitude and serum LH levels generally are somewhat lower in obese than in lean women with PCOS.7,60 Elevated serum LH concentrations in women with PCOS also exhibit increased bioactivity in bioassay systems in vitro, reflecting a difference in glycosylation with a predominance of more basic (alkaline) LH isofoms, which have greater bioactivity.53,61,62 and 63
The approximate hourly LH pulse frequency in women with PCOS is within the range of frequencies usually observed across the normal ovulatory cycle, suggesting it results from a failure of the mechanisms that normally slow the GnRH pulse generator rather than from an abnormal acceleration in pulse frequency. The increased pulse frequency might reflect intrinsic hypothalamic dysfunction, the effects of abnormal feedback signals from the periphery, or both.64
Since dopamine and opioids normally inhibit hypothalamic GnRH neuronal activity, the higher GnRH pulse frequency observed in women with PCOS could be caused by a decrease in dopaminergic or opioidergic neuronal stimulation. However, experimental evidence from studies involving treatment with medications that stimulate or inhibit these pathways does not support either mechanism. Treatment with a dopamine agonist has no discernible effect on the pattern of gonadotropin secretion in women with PCOS.65,66 Treatment with a progestin slows LH pulse frequency,9 just as progesterone does during the normal luteal phase, indicating that the opioid-dependent process that normally mediates the effects of progesterone is operating,67,68 and 69 and suggesting that any decrease in opioid tone results primarily from the lack of progesterone feedback, due to anovulation.
Infusion of exogenous insulin70,71 and 72 and treatments that decrease insulin levels (metformin, thiazoladinediones) have no significant effect on the pattern of LH secretion in women with PCOS.72,73 LH levels also are lower in obese than in lean women with PCOS, even though insulin levels are higher in the obese.7,60 These observations suggest that hyperinsulinemia has no significant direct effect on LH secretion.
Treatment with exogenous estrone does not increase basal or GnRH-stimulated LH concentrations in women with PCOS,74 and treatment with an aromatase inhibitor does not decrease LH pulse frequency,75 indicating that increased circulating levels of estrone may exert negative feedback effects on FSH, but probably do not have any important direct influence on LH secretion in women with PCOS. Whereas the lack of progesterone feedback resulting from anovulation undoubtedly contributes to the higher LH pulse frequency,9 evidence suggests that the GnRH pulse generator also is less sensitive to the feedback inhibition of sex steroids.
Treatment with an estrogen-progestin contraceptive or with physiologic doses of exogenous estrogen and progesterone slows LH pulse frequency in women with PCOS, but to a lesser extent than in normal women.76,77 and 78 However, after pretreatment with flutamide (an androgen receptor antagonist), the effects of estrogen and progesterone on LH pulse frequency in women with PCOS are the same as in normal women,10 suggesting that increased circulating androgen levels help to sustain the higher LH pulse frequency observed in women with PCOS by decreasing sensitivity to estrogen and progestin feedback.
Androgens also may contribute more directly to the abnormal pattern of gonadotropin secretion in women with PCOS. Evidence from studies in rats, sheep, monkeys, and women indicates that prenatal exposure to increased androgen concentrations may affect GnRH pulse generator programming, predisposing to an increased pulse frequency and LH secretion.12,13,79,80,81 and 82 At least in rodents, prenatal androgen treatment also decreases basal and estrogen-induced hypothalamic progesterone receptor concentrations,81 offering a mechanism to explain how androgens might decrease hypothalamic sensitivity to progesterone feedback. It could be that hyperandrogenemia from any cause, arising during fetal life (maternal hyperandrogenism, classical congenital adrenal hyperplasia), adolescence (premature adrenarche, nonclassical congenital adrenal hyperplasia), or in adulthood (obesity, hyperinsulinemia) induces abnormalities in the feedback control of pulsatile GnRH secretion, resulting in increased LH secretion, which stimulates increased ovarian androgen production, in a self-perpetuating cycle.
The primary evidence indicating that excessive LH stimulation plays an important role in the pathophysiology of PCOS comes from studies examining the effects of treatment with GnRH antagonists and long-acting GnRH agonists. In women with PCOS, treatment with a GnRH antagonist induces an acute dose-dependent decrease in both LH and testosterone concentrations,54 and long-term treatment with an agonist can suppress ovarian androgen production to postmenopausal levels.83,84 However, normally cycling women with polycystic ovaries exhibit higher androgen and insulin levels and lower SHBG concentrations than women with normal ovarian morphology, even though LH levels and secretory dynamics are not different.85 These observations suggest that excessive LH secretion or stimulation may be an important cause of disordered follicular development and anovulation, but is not the proximate cause of polycystic ovaries or of increased ovarian androgen production in women with PCOS.
Insulin Secretion and Action
An association between glucose intolerance and hyperandrogenism was first recognized by Archard and Thiers in 1921, in a famous report describing a bearded diabetic woman.86 Insulin resistance was first described in diabetic patients who required progressively higher doses of insulin to maintain effective glucose control, most often because they developed antibodies to preparations of insulin derived from animal sources.87 Today, we recognize insulin resistance as a feature of a wide variety of disorders and conditions, ranging from extreme insulin-resistance syndromes (auto-antibodies to the insulin receptor, insulin receptor mutations, lipodystrophic states)88,89 and 90 to common problems such as type 2 diabetes, obesity, stress, infection, pregnancy, and PCOS. The importance of insulin resistance, hyperinsulinemia, and insulin action in the pathogenesis of PCOS was first suggested by a study conducted in 1980, demonstrating significant correlations between basal levels of plasma insulin, androstenedione, and testosterone, and between insulin and testosterone levels after an oral glucose load.91
Insulin resistance is a common feature in obese and, to a lesser extent, lean women with PCOS; the overall prevalence ranges between 50% and 75%.92,93,94 and 95 Insulin sensitivity is decreased by an average of 35-40% in women with PCOS, compared to normal
women, similar to what is observed among women with noninsulin dependent diabetes mellitus.96,97 and 98 Up to 35% of women with PCOS exhbit impaired glucose tolerance and 7-10% meet criteria for type 2 diabetes mellitus.99,100 Conversely, women with type 2 diabetes are 6-fold more likely than non-diabetic women of similar age and weight to have PCOS.101
women, similar to what is observed among women with noninsulin dependent diabetes mellitus.96,97 and 98 Up to 35% of women with PCOS exhbit impaired glucose tolerance and 7-10% meet criteria for type 2 diabetes mellitus.99,100 Conversely, women with type 2 diabetes are 6-fold more likely than non-diabetic women of similar age and weight to have PCOS.101
Insulin resistance is a condition in which endogenous or exogenously administered insulin has less than normal effects on fat, muscle, and the liver.102 In adipose, insulin resistance results in increased hydrolysis of stored triglycerides and elevated circulating free fatty acid levels. Decreased glucose utilization (primarily in muscle) and increased hepatic gluconeogenesis (which insulin normally inhibits) result in increased blood glucose concentrations and a compensatory hyperinsulinemia (in those with adequate pancreatic reserve). Increased circulating insulin levels cause or contribute to hyperandrogenism in women with PCOS in at least two important ways, by stimulating increased ovarian androgen production, and by inhibiting hepatic SHBG production.
Numerous studies have demonstrated that insulin stimulates androgen production in ovarian theca cells in vitro.103 Theca cells from women with PCOS also exhibit increased sensitivity to insulin, compared to those from normal women. Physiologic levels of insulin can stimulate androgen synthesis in theca cells of women with PCOS, whereas higher insulin concentrations are required in normal theca cells.104,105 Because insulin also potentiates the action of LH,106 insulin and LH act synergistically to stimulate androgen production.104,107
Clinical investigations in women with PCOS have demonstrated that insulin also stimulates ovarian androgen production in vivo. Notably, the cumulative sum insulin response during an oral glucose tolerance test correlates positively with the rise in serum androstenedione and testosterone above baseline concentrations.108 Moreover, suppression of serum insulin
levels by treatment with diazoxide or an insulin-sensitizing agent (troglitazone) decreases serum androstenedione and testosterone levels in women with PCOS.109,110
levels by treatment with diazoxide or an insulin-sensitizing agent (troglitazone) decreases serum androstenedione and testosterone levels in women with PCOS.109,110
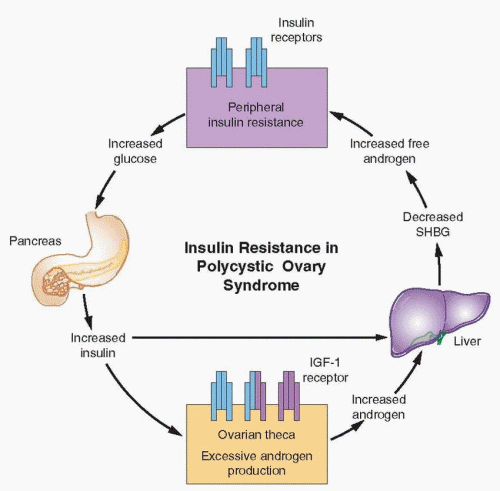 |
High insulin concentrations also inhibit hepatic SHBG production,111,112 as do high androgen concentrations. The combined actions of insulin and androgens lower SHGB concentrations, yielding increased free androgen levels, which aggravate the underlying insulin resistance.113 Ultimately, these conditions foster a self-propagating positive feedback loop that can increase in severity over time.
Insulin stimulates ovarian androgen production acting via insulin receptors on theca/ interstitial cells in the ovarian stroma.105,114 At high concentrations, insulin also binds to IGF-1 receptors (and possibly hybrid receptors) which are structurally similar and use a similar signaling mechanism.96 However, evidence indicates that insulin acts primarily via its own receptor, by activating a signaling system separate from that involved in glucose transport. Whereas an anti-insulin receptor antibody effectively inhibits insulin-stimulated steroidogenesis in cultured human granulosa cells, an anti-IGF receptor antibody has no effect.105,115,116 A d-chiro-inositol containing glycan increases theca cell testosterone production in vitro, and preincubation with an anti-inositolglycan antibody blocks insulin stimulation, but not that of hCG.105 These observations suggest inositolphosphoglycan mediators act as second messengers in signal transduction for insulin stimulation of theca cell androgen synthesis and that the mechanism differs from that mediating the actions of LH.117
What causes insulin resistance in women with PCOS is not entirely clear. Not surprisingly, given the complexity and polygeneic nature of the disorder, evidence suggests that more than one mechanism may be involved.
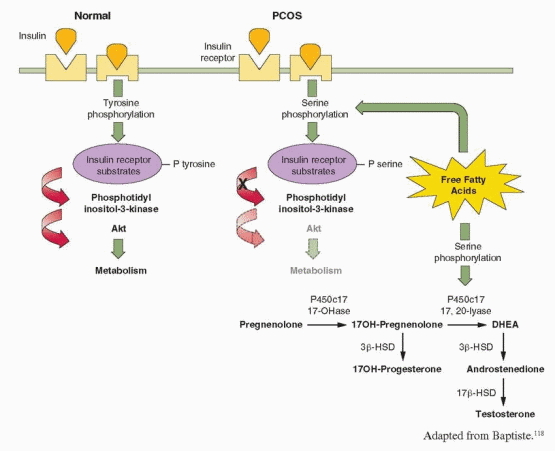
The classical actions of insulin are mediated via its receptor and two distinct intracellular pathways. The phosphatidyl-inositol 3-kinase (PI-3K) pathway mediates the metabolic effects of insulin, and the mitogen-activated protein kinase (MAPK) pathway mediates the proliferative actions of insulin. Normally, insulin binding to its receptor induces a conformational change, resulting in tyrosine phosphorylation of the receptor and protein substrates, which bind and serially activate PI-3K and Akt, an effector molecule that plays the major role in signal transduction for glucose regulation and metabolism.118,119 Akt activation potentiates the translocation of glucose transporter 4 (GLUT4) from intracellular compartments to the plasma membrane, thereby increasing glucose uptake. Other effector molecules mediate insulin inhibition of gluconeogenesis and glycogenolysis,120,121 stimulation of lipid synthesis, and inhibition of lipid catabolism.122,123 Luteinized granulosa cells obtained from women with PCOS display both a selective increase in insulin activation of the mitogenic pathway, via MAPK, and resistance in the PI-3K-mediated metabolic pathway of insulin action.124 These and similar observations illustrate how insulin actions can be selectively inhibited and enhanced at the same time, via different signaling pathways,96,124 explaining how insulin can stimulate hyperandrogenism in women who are “insulin-resistant.”
Studies in cultured skin fibroblasts, muscle, and adipocytes from women with PCOS indicate that insulin resistance results from defects early in the postreceptor signaling pathway.98,125,126 and 127 The number and affinity of insulin receptors in both obese and lean women with PCOS are not decreased,128,129 but insulin receptors exhibit a constitutive increase in phosphorylation of serine residues and a decrease in insulin-stimulated phosphorylation of tyrosine residues. Serine phosphorylation of insulin receptor substrates prevents their binding with PI-3K and thereby inhibits insulin signaling. Increased serine phosphorylation can be induced by intracellular metabolites of free fatty acids,126,130 which are increased in most women with PCOS and have been demonstrated to cause insulin resistance in vivo.131,132 High circulating free fatty acid levels also can increase androgen production in women,133 by inducing serine phosphorylation of P450c17, which results in increased 17,20 lyase
activity.134,135 These observations offer a mechanism for insulin resistance that also further helps to explain the link between insulin and hyperandrogenism in women with PCOS.
activity.134,135 These observations offer a mechanism for insulin resistance that also further helps to explain the link between insulin and hyperandrogenism in women with PCOS.
 |
Although hyperandrogenism can decrease insulin sensitivity, the effect is relatively modest.113 Insulin resistance and hyperinsulinemia are the primary factors; they are the cause, not the result, of hyperandrogenism. Treatment with a GnRH agonist can normalize elevated serum androstenedione and testosterone levels in women with PCOS, but has limited or no effect on insulin resistance.37,136,137 and 138 Similarly, although bilateral ovarian cautery can decrease serum androgen concentrations by nearly 50% in women with PCOS, glucose utilization (insulin sensitivity) remains unchanged.139
Accumulating evidence suggests that deficiency or dysfunction in downstream signaling mediated by inositolphosphoglycans also may contribute to insulin resistance in women with PCOS.140,141,142 and 143 Finally, obesity is a common feature of women with PCOS, representing yet another important mechanism contributing to the development of insulin resistance, as discussed below.
Insulin resistance and hyperinsulinemia are undoubtedly an important part of the pathophysiology of PCOS. However, it is important to emphasize that 25-50% of women with PCOS have no demonstrable insulin resistance. Moreover, among all women with insulin resistance, the prevalence of PCOS is relatively low (approximately 15%).144 Therefore, insulin resistance and hyperinsulinemia are not the primary cause or pathogenic factor in all women with PCOS.
Weight and Energy Regulation
The risk for developing PCOS rises with increasing obesity,144,145 and 146 as does the severity of insulin resistance, hyperinsulinemia, and ovulatory dysfunction, and the prevalence of metabolic syndrome, glucose intolerance, risk factors for cardiovascular disease, and sleep apnea.100, 147-150
Obesity, by itself, is associated with insulin resistance and compensatory hyperinsulinemia. Insulin resistance is most highly correlated with intra-abdominal obesity, because visceral fat is more active metabolically than subcutaneous fat, more sensitive to lipolysis, releases more free fatty acids, and produces a number of cytokines involved in insulin resistance, such as tumor necrosis factor-α (TNF-α), interleukin-6, leptin, and resistin.151 The accumulation of free fatty acids in tissues causes lipotoxicity and insulin resistance, in part via TNF-α, which increases serine phosphorylation and thereby inhibits insulin signaling.152 Insulin resistance due to obesity also induces leptin resistance and decreases adiponectin levels, thereby decreasing fatty acid oxidation and promoting lipotoxicity.151,153 Obesity in women with PCOS typically is distributed centrally, with a greater increase in visceral than in subcutaneous fat.154,155,156 and 157 However, even lean women with PCOS have an increased percentage of body fat, a higher waist-hip ratio, and greater intra-abdominal, peritoneal and visceral fat, compared to normal women matched for body mass index (BMI).
The overall prevalence of obesity, and in women with PCOS, varies among different patient populations;158 in the United States, approximately 35% of all adult women and 60% of women with PCOS are obese.159,160 However, the overall prevalence of PCOS among different populations is quite similar (approximately 7%).30,161,162 and 163 Moreover, the prevalence of PCOS among unselected women varies relatively little with increasing BMI: 8.2% in underweight women (BMI < 18.5), 9.8% in normal-weight women, 9.9% in overweight women (BMI 25.0-30.0), 9.0% in obese women (BMI ≥ 30.0), 12.4% in those with a BMI between 35.0 and 40.0, and 11.5% in morbidly obese women (BMI > 40.0).146 Combined, these observations indicate that obesity relates primarily to genetic and environmental factors and is a common, but not essential, feature of PCOS. Obesity contributes modestly to the risk for developing PCOS and adds to the pathophysiology in already affected women by aggravating the degree of insulin resistance and hyperinsulinemia.164,165 It also is possible that PCOS itself may, to some degree, predispose to weight gain and obesity.
The prevalence of menstrual irregularity, dysfunctional bleeding, hirsutism, and infertility is higher in obese than in lean women with PCOS,165,166 and 167 as is the risk for developing glucose intolerance and diabetes.100,168 Moreover, obese women have a higher prevalence of miscarriage, gestational diabetes, and pre-eclampsia, regardless whether they also have PCOS.169
Androgen Synthesis and Action
Hyperandrogenism is the key feature of PCOS, resulting primarily from excess androgen production in the ovaries and, to a lesser extent, in the adrenals.170 In women with PCOS, approximately 60% of circulating androstenedione derives directly from the ovaries and the remainder from the adrenals; similarly, 60% of circulating testosterone is secreted directly by the ovaries, with most of the remainder deriving from peripheral conversion of androstenedione.171
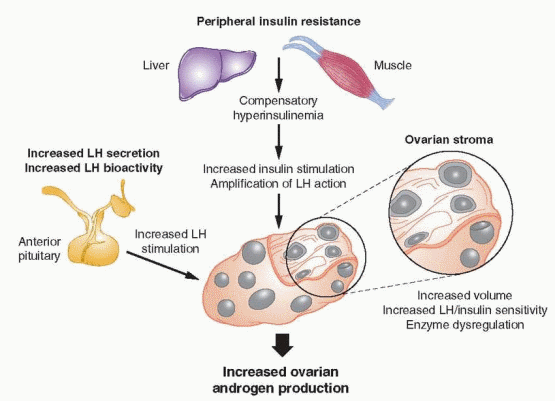
The primary mechanisms driving increased ovarian androgen production in PCOS include increased LH stimulation resulting from abnormal LH secretory dynamics and
increased LH bioactivity, and hyperinsulinemia due to insulin resistance, which potentiates the action of LH and is worsened by obesity. Other evidence indicates that increased ovarian androgen synthesis in women with PCOS probably also relates to the increased volume of theca cells in an expanded ovarian stroma, and to increased sensitivity to LH stimulation,172,173 possibly due to over-expression of LH receptor in theca and interstitial (stromal) cells.174 Androgen production remains elevated in long-term cultures of theca cells from women with PCOS,175 even after suppression of LH levels by treatment with a GnRH agonist,176 suggesting that increased androgen production in women with PCOS, to some extent, also reflects an intrinsic dysregulation of key steroidogenic enzmes, such as 3β-hydroxysteroid dehydrogenase (3β-HSD) and 17,20-lyase,104,172,177,178 and 179 which may have a genetic foundation.180
increased LH bioactivity, and hyperinsulinemia due to insulin resistance, which potentiates the action of LH and is worsened by obesity. Other evidence indicates that increased ovarian androgen synthesis in women with PCOS probably also relates to the increased volume of theca cells in an expanded ovarian stroma, and to increased sensitivity to LH stimulation,172,173 possibly due to over-expression of LH receptor in theca and interstitial (stromal) cells.174 Androgen production remains elevated in long-term cultures of theca cells from women with PCOS,175 even after suppression of LH levels by treatment with a GnRH agonist,176 suggesting that increased androgen production in women with PCOS, to some extent, also reflects an intrinsic dysregulation of key steroidogenic enzmes, such as 3β-hydroxysteroid dehydrogenase (3β-HSD) and 17,20-lyase,104,172,177,178 and 179 which may have a genetic foundation.180
Adrenal androgen production (androstenedione, DHEA, DHEA-S) also is increased in women with PCOS; over half exhibit moderately increased circulating DHEA-S levels.181 When ovarian synthesis is suppressed by treatment with a long-acting GnRH agonist, adrenal androgen levels remain higher in women with PCOS than in normal women.92,172,176,182 Adrenal androgens have little or no intrinsic androgenic activity, but contribute to the pathophysiology of PCOS via conversion to testosterone in the periphery.
A number of potential mechanisms for the increase in adrenal androgen production have been investigated, but the explanation remains uncertain. Chronic estrogen stimulation due to anovulation could decrease adrenal 3β-HSD activity, as it does in the fetal adrenal cortex, but evidence for the mechanism is conflicting.84,183,184,185 and 186 Increased pituitary ACTH secretion or increased sensitivity to ACTH could provide an explanation, but neither can be demonstrated.181,187,188 In some, but clearly not all women with PCOS, adrenal androgen excess might result from intrinsic upregulation of P450c17 17,20 lyase activity,39,135,189,190 and 191 or from hyperinsulinemia.192,193,194,195 and 196 In sum, no one mechanism explains the moderate adrenal androgen excess commonly observed in women with PCOS.
High local androgen concentrations contribute to the polycystic morphogenesis of the ovaries, via conversion to more potent 5α-reduced androgens, which cannot be aromatized to estrogen and inhibit both aromatase activity and FSH induction of LH receptors on granulosa cells, thereby impeding or preventing progressive follicular development. Granulosa cells obtained from polycystic ovaries are not functionally impaired. They are sensitive to FSH and insulin-like growth factors and produce estrogen,197,198,199,200 and 201 but cannot generate and maintain the estrogenic follicular milieu required to achieve more advanced stages of development. Consequently, new follicular growth continues but arrests long before full maturation is achieved, resulting in multiple small follicular cysts (typically measuring 2-10 mm in diameter), surrounded by hyperplastic theca cells, which often become luteinized due to increased LH stimulation. Atretic follicles ultimately contribute to an expanding ovarian stroma that increases in volume over time, further increasing the cellular mass producing androgens, in yet another self-propagating cycle that predisposes to chronic anovulation.
The importance of high local ovarian androgen concentrations in the pathophysiology of PCOS is demonstrated by the results of ovarian wedge resection and by observations in women with other conditions associated with hyperandrogenemia. Wedge resection results in a sustained decrease in androgen levels that precedes the return of ovulatory cycles, indicating that high intraovarian androgen concentrations effectively inhibit follicular development and prevent ovulation.202,203,204 and 205 The success of ovarian wedge resection correlates with the amount of androgen-producing stromal tissue that is removed; even a unilateral oophorectomy can restore menstrual cyclicity and ovulation in anovulatory women with polycystic ovaries.206 Although laparoscopic procedures such as ovarian “drilling” with an electrosurgical needle or a laser have replaced the classical wedge resection, the results achieved are similar. Polycystic ovaries also have been observed in women with androgen-producing ovarian and adrenal tumors,207,208 and 209 and in female-to-male transsexuals
treated with exogenous androgens.210,211 These observations again illustrate the important point that polycystic ovaries are not a characteristic feature of a specific endocrine disorder. They result from a functional derangement in follicular development induced or sustained by increased intraovarian androgen levels as a consequence of chronic anovulation, whatever the cause.
treated with exogenous androgens.210,211 These observations again illustrate the important point that polycystic ovaries are not a characteristic feature of a specific endocrine disorder. They result from a functional derangement in follicular development induced or sustained by increased intraovarian androgen levels as a consequence of chronic anovulation, whatever the cause.
 |
Genetic Considerations
Familial clustering of hyperandrogenism, anvoulation, and polycystic ovaries suggests an underlying genetic basis or cause. At least one group of patients with a heritable X-linked form of PCOS has been described, albeit with a widely varying phenotype.212 Studies in large families have suggested autosomal-dominant inheritance, with premature balding as the male phenotype.213,214 Other studies of siblings and parents of women with PCOS have observed a high prevalence of hyperinsulinemia and hypertriglyceridemia, PCOS in females, and premature balding in males.215,216 Nearly 50% of sisters of women with PCOS have elevated total or bioavailable testosterone concentrations,217 and approximately 35% of mothers also are affected.218,219 The first degree relatives of women with PCOS also exhibit other metabolic abnormalities such as dyslipidemia, which may predispose to an increased risk for cardiovascular disease.220,221,222 and 223 These observations further suggest a genetic predisposition or susceptibility.
Understandably, efforts to identify genes associated with a susceptibility to anovulation and polycystic ovaries have focused on genes relating to the insulin receptor and substrates224,225 and 226 and the genes encoding the P450 side-chain cleavage (CYP11) and P450c17 (CYP17) enzymes.227,228,229,230 and 231 However, it seems likely that PCOS is a polygenic disorder involving the interaction of numerous genomic variants and the influence of environmental factors.232 Candidate genes include the long list of molecules that participate in any of the metabolic and reproductive pathways affected in the syndrome, emphasizing yet again that PCOS is not a specific endocrine disorder, but a result of chronic anovulation due to a wide variety of causes.
Summary of Key Points
Polycystic ovary syndrome is not a specific endocrine disorder having a unique cause. Rather, it is a complex disorder wherein numerous genetic variants and environmental factors interact, combine, and contribute to the pathophysiology.
Polycystic ovaries and the clinical features of polycystic ovary syndrome reflect a functional derangement in follicular development, resulting in chronic anovulation. Because there are many causes of anovulation, there are many causes of polycystic ovaries and the polycystic ovary syndrome.
Women with polycystic ovary syndrome generally exhibit increased serum LH concentrations, low-normal FSH levels, and increased LH:FSH ratios. The increase in serum LH levels results from abnormal LH secretory dynamics, characterized by increases in LH pulse frequency and amplitude, reflecting the pattern of pulsatile GnRH secretion. The decrease in FSH levels results from the increase in GnRH pulse frequency and from the negative feedback of chronically elevated estrone concentrations (derived from peripheral aromatization of increased androstenedione) and normal or increased levels of inhibin B (derived from small follicles).
Insulin resistance and compensatory hyperinsulinemia are common features in women with polycystic ovary syndrome and play an important role in the pathophysiology. Up to 35% of women with polycystic ovary syndrome exhibit impaired glucose tolerance and up to 10% meet criteria for type 2 diabetes mellitus.
Increased LH and insulin stimulation drives ovarian androgen production, and androgens and insulin combine to inhibit hepatic SHBG production, yielding increased free androgen, which aggravates underlying insulin resistance, in a self-propagating positive feedback loop that can increase in severity over time.
Obesity contributes to the risk for developing polycystic ovary syndrome and adds to the pathophysiology in already affected women by aggravating the degree of insulin resistance and hyperinsulinemia.
Hyperandrogenism is a major feature of polycystic ovary syndrome, resulting primarily from excess androgen production in the ovaries and, to a lesser extent, in the adrenals. Increased LH and insulin stimulation are the primary mechanisms driving increased ovarian androgen production. Others include an expanded ovarian stoma having increased sensitivity to insulin and LH, and intrinsic dysregulation of key steroidogenic enzymes.
Polycystic ovary syndrome is a polygenic disorder likely involving the interaction of numerous genomic variants and the influence of environmental factors. Candidate genes include all of the molecules that participate in the affected metabolic and reproductive pathways.
Diagnosis of Polycystic Ovary Syndrome
It is generally accepted that PCOS is not a specific endocrine disease but a syndrome represented by a collection of signs and symptoms, and that no one sign, symptom, or test is diagnostic. Not surprisingly, establishing criteria for diagnosis of PCOS has proven both challenging and controversial.
It has been argued that having a clear and specific definition for PCOS is important because affected women are at increased risk for a variety of problems (infertility, dysfunctional bleeding, endometrial cancer, obesity, type 2 diabetes mellitus, dyslipidemia, hypertension, and cardiovascular disease), because the diagnosis can have health implications for other family
members, and because the need for life-long treatment may affect access to health care insurance in systems like that in the United States.43,233 In our view, the primary advantage to having specific diagnostic criteria for PCOS relates to research, because varying criteria cloud the conclusions and question the generalizability of results from studies involving women with “PCOS.” In clinical medicine, simply knowing and understanding the health implications and consequences of chronic anovulation and methods for their effective management are far more important than assigning a specific diagnosis of PCOS.
members, and because the need for life-long treatment may affect access to health care insurance in systems like that in the United States.43,233 In our view, the primary advantage to having specific diagnostic criteria for PCOS relates to research, because varying criteria cloud the conclusions and question the generalizability of results from studies involving women with “PCOS.” In clinical medicine, simply knowing and understanding the health implications and consequences of chronic anovulation and methods for their effective management are far more important than assigning a specific diagnosis of PCOS.
The basis for diagnosis of PCOS has changed with time and advances in medicine and related technology. The earliest descriptions of the disorder were based on findings of enlarged ovaries, hirsutism, and menstrual dysfunction.32 The advent of hormone assays moved the focus to serum gonadotropin and androgen concentrations.234 More recent advances in ultrasonography and recognition of the importance of insulin resistance in the pathophysiology have turned attention to ovarian morphology235 and to the metabolic consequences of the disorder.
There have been three separate and distinct efforts to establish or refine the diagnostic criteria for PCOS. The first was a conference sponsored by the National Institute of Child Health and Human Development (NICHD) in 1990, concluding that the major criteria for diagnosis of PCOS (in order of importance) were (1) hyperandrogenism and/or hyperandrogenemia, (2) menstrual dysfunction, and (3) exclusion of other known disorders having a similar clinical presentation.233 The second was a conference co-sponsored by the European Society for Human Reproduction and Embryology (ESHRE) and the American Society for Reproductive Medicine (ASRM), convened in Rotterdam, The Netherlands, in 2003, concluding that diagnosis of PCOS should be based on at least two of three major criteria, including (1) oligo/anovulation, (2) clinical or biochemical signs of hyperandrogenism, and (3) polycystic ovaries (as identified by ultrasonography), also excluding other androgen excess disorders.236,237 The third was a task force appointed by the Androgen Excess and PCOS Society (AE-PCOS) in 2006, concluding that diagnosis of PCOS requires (1) hyperandrogenism (hirsutism and/or hyperandrogenemia), (2) ovarian dysfunction (oligo/anovulation and/or polycystic ovaries), and (3) exclusion of other androgen excess or related disorders.43
The original 1990 NICHD diagnostic criteria were based on traditional concepts of PCOS, requiring evidence of both hyperandrogenism (hyperandrogenemia and/or hirsutism) and menstrual dysfunction (oligo/amenorrhea). The 2003 ESHRE/ASRM (“Rotterdam”) criteria sought to recognize and accommodate a broader spectrum of the disorder, regarding polycystic ovaries as evidence of ovarian dysfunction and including women having neither hyperandrogenemia nor hirsutism. The 2006 AE-PCOS Society criteria allowed that polycystic ovaries could be considered a sign of ovarian dysfunction, but again emphasized that PCOS is characterized, first and foremost, by hyperandrogenism, including women with either oligo/amenorrhea or polycystic ovaries, but excluding those having neither hyperandrogenemia nor hirsutism.
Ironically, although the purpose of the consensus conferences and task force was to rigorously define PCOS for purposes of research, it can be argued that the differing sets of criteria succeeded only in creating controversy and confusion where clarity was needed most. Published clinical trials involving women with PCOS must be carefully reviewed to determine which diagnostic criteria were applied in selecting the study population.
Hyperandrogenemia
Biochemical evidence of hyperandrogenism is based on the finding of elevated circulating androgen concentrations. Testosterone is the most important androgen produced by the
ovary and the usual basis for diagnosis of hyperandrogenemia. Other androgens that may be elevated in women with PCOS include androstenedione, DHEA, and DHEA-S.
ovary and the usual basis for diagnosis of hyperandrogenemia. Other androgens that may be elevated in women with PCOS include androstenedione, DHEA, and DHEA-S.
Testosterone levels are elevated in most, but not all, women with PCOS. The free testosterone level is more sensitive for diagnosis of hyperandrogenic disorders,43 but measurements of free testosterone have several limitations. Direct radioimmunoassays (RIA) for free testosterone are highly inaccurate,238,239 and 240 particularly in the lower range and in women with decreased SHBG levels.241 More sophisticated and accurate methods (equilibrium dialysis, gas or liquid chromatography-mass spectrometry) are technically complex, costly, and not widely available.242 Moreover, testosterone is converted in androgen-sensitive tissues to dihydrotestosterone (DHT), which has a longer duration of action than testosterone, so serum total testosterone concentrations do not necessarily reflect androgen bioactivity.
For purposes of clinical research, the free testosterone concentration can be calculated, using equations derived from the laws of mass action, the serum concentrations of total testosterone, SHBG, and albumin, and the association constants for the interactions of testosterone with SHBG and albumin.239,243 Calculated values generally correlate well with those determined by equilibrium dialysis,238,239 although accuracy varies with the specific assays used to measure total testosterone and SHBG. For clinical purposes, measurement or calculation of the free testosterone level, or even measurement of the serum total testosterone concentration, usually is unnecessary.244 In most cases, hirsutism provides ample evidence of hyperandrogenism, and if not severe, sudden in onset, rapidly progressive, or associated with symptoms or signs of virilization, there is little reason for concern about an androgen-producing tumor (Chapter 13).
Measurement of the serum androstenedione concentration could yield evidence of hyperandrogenemia, but limited data suggest that levels are elevated in less than 20% of women with PCOS.29 Measurement of serum DHEA also has little or no diagnostic value because levels are relatively low, exhibit a diurnal pattern and high between-subject variability, and are sensitive to stress.43,245
The serum DHEA-S concentration is the traditional marker for adrenal androgen excess,246,247 and 248 because it derives almost exclusively from the adrenal,249,250 and 251 and concentrations are relatively high and remain stable across the day and cycle.252,253 Overall, the serum DHEA-S concentration is moderately elevated in over half of women with PCOS.181 Some have an isolated increase in serum DHEA-S, suggesting a deficiency in 3β-HSD, but no genetic mutation in the enzyme has been found.254,255 Although the AE-PCOS Society regards an elevated serum DHEA-S level as sufficient evidence of hyperandrogenism to support the diagnosis of PCOS,43 the test has very limited or no clinical value in our view. First, the test lacks both sensitivity and specificity for identifying women with adrenal causes of hyperandrogenism.256 Second, DHEA-S, like DHEA and androstenedione, has little or no intrinsic androgenic activity and requires conversion to testosterone to exert androgenic effects. Third, the DHEA-S concentration can be grossly elevated (≥700 μg/dL) in women with rare androgen-secreting tumors, but in almost all such patients, the serum testosterone level also is greatly elevated,257 due to peripheral conversion of high circulating DHEA-S levels, or because the tumor also secretes testosterone.
Clinical Hyperandrogenism
Clinical evidence of hyperandrogenism includes hirsutism, acne, and androgenic alopecia, all of which relate to the effect of androgens on the pilosebaceous unit. Because the sensitivity of the pilosebaceous unit varies significantly among individuals, the correlation between these clinical features and biochemical measures of hyperandrogenism is relatively poor.258,259
Hirsutism is the growth of terminal hairs on the face or body in a male pattern. Hirsutism is the most obvious clinical indicator of androgen excess and is an important feature of PCOS. Whereas hirsutism affects 65-75% of White, Black and Southeast Asian women,43,260 its prevalence is lower in racial or ethnic groups having relatively little body hair.260,261 and 262 The modified Ferriman-Gallwey score is the most common method for grading the extent of hirsutism, assigning a score from 0-4 in each of 9 androgen-sensitive areas, as illustrated and described in Chapter 13.263,264 The threshold value that defines hirsutism is not firmly established, but generally has ranged between 6 and 8.29,260,264 The modified Ferriman-Gallwey score is the accepted standard for assessing the severity of hirsutism in clinical investigations. However, in clinical practice, the easiest and most practical way is to determine the method and frequency of hair removal (e.g., shaving, plucking, waxing), which also provides a clinically relevant measure for assessing the response to treatment.
Acne can be another manifestation of hyperandrogenism. Like hirsutism, its prevalence among women with PCOS varies with ethnicity. The prevalence of acne is 12-14% among White women with PCOS,159,161,262 higher in Asian Indians and women of Mediterranean descent (approximately 25%),262,265 and lower among Pacific Islanders.261 However, it is unclear whether acne is any more prevalent among women with PCOS than in the general population. Approximately 20% of women under age 20, 15% of those ages 20 to 30, and 10% of women ages 30 to 40 complain of acne.266,267,268,269 and 270 The extent to which PCOS may increase risk for developing acne, if at all, is therefore uncertain.
Androgenic alopecia, describing scalp hair loss in women, also can result from hyperandrogenism and is a recognized, but uncommon, feature of PCOS;23,159,271,272 and 273 less than 5% of women with PCOS complain of hair loss. Typically, the hair loss is limited to the crown and does not involve the frontal hair line.272,274 Androgenic alopecia may be more common than is recognized, because 25% or more of scalp hair must be lost before thinning becomes apparent.159,274
Ovulatory and Menstrual Dysfunction
Normal cyclic menses result from normal ovulatory function. The normal inter-menstrual interval ranges between 24 and 35 days and menses that occur less or more often are an indication of ovulatory dysfunction. Cyclic menses occurring at normal intervals strongly suggest, but cannot be regarded as proof of ovulation.
The majority of women with PCOS, approximately 60-85%, exhibit gross menstrual dysfunction.43,158,161 The most common abnormalities are oligomenorrhea and amenorrhea. Polymenorrhea (regular cycles occurring at intervals less than 25 days) is very uncommon, observed in less than 2% of untreated women with PCOS.159 Classically, menstrual dysfunction in women with PCOS has a premenarcheal onset, but many report regular cycles for varying intervals preceding the onset of oligo/amenorrhea.
In general, anovulatory women seldom have regular menses.275 However, regular cycles are somewhat more common in anovulatory hyperandrogenic women.43,276 In studies of menstrual function in women with hyperandrogenism, approximately 15-40% are eumenorrheic, despite evidence of oligo-anovulation.277,278,279 and 280 The prevalence of eumenorrhea among women with PCOS is significantly increased if the Rotterdam diagnostic criteria are applied, because hirsute eumenorrheic women with polycystic ovaries are included. The absence of any recognizable pattern of premenstrual molimina suggests anovulation in eumenorrheic women.
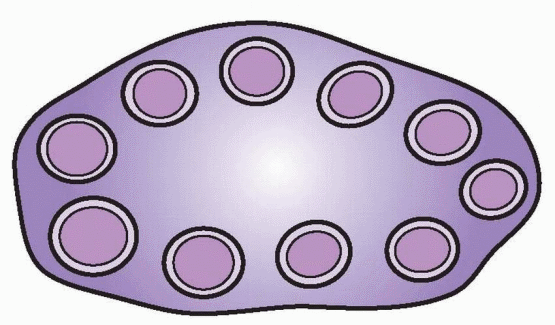
Polycystic Ovaries
PCOS takes its name from the enlarged polycystic ovaries so commonly observed in women with hyperandrogenic chronic anovulation.32 Observations of mild hyperandrogenemia and insulin resistance in some asymptomatic women with polycystic ovaries provided the rationale for including polycystic ovaries among the Rotterdam diagnostic criteria for PCOS, as a sign of ovarian dysfunction.85,281,282,283 and 284
Polycystic ovaries typically exhibit increased size and stromal volume and an increased number of small follicles. The Rotterdam criteria consider only the total number of follicles, requiring 12 or more measuring 2-9 mm in diameter (mean of both ovaries).236,237,285,286 and 287 Others have defined polycystic ovaries on the basis of volume (>7.0-7.5 mL) and architecture.288,289 The prevalence of polycystic ovaries is quite high among women with androgen excess (>80%).235,277,290,291,292,293,294 and 295 However, from 8% to 25% of normal women, and even 14% of women using oral contraceptives, also meet the ultrasonographic criteria for polycystic ovaries.281, and 296,297,298 and 299 300 Moreover, polycystic ovaries are commonly observed during normal pubertal development, and even in women with hypothalamic amenorrhea and hyperprolactinemia.301,302
The 2003 Rotterdam diagnostic criteria expanded the population of women that might be assigned a diagnosis of PCOS by approximately 50%, compared to the criteria earlier recommended by the NICHD, due entirely to the inclusion of polycystic ovaries.303 The change in criteria ignited considerable controversy, primarily because polycystic ovaries are so commonly observed in normal women and in other conditions. Moreover, the finding, by itself, has little clinical significance. Otherwise normal women with polycystic ovaries generally have regular menstrual cycles, exhibit normal serum gonadotropin and ovarian steroid hormone levels, and are not infertile.85,297,304,305 and 306
Other Features of the Polycystic Ovary Syndrome
PCOS has other common features besides hyperandrogenism and ovulatory dysfunction that are not included in any diagnostic criteria, including abnormal patterns of gonadotropin secretion, insulin resistance, and related metabolic abnormalities, such as dyslipidemia.
Abnormal Gonadotropin Secretion
Abnormal patterns of gonadotropin secretion have long been recognized as a common characteristic of women with PCOS. As discussed earlier in the section of this chapter devoted to the pathophysiology of the disorder, increased serum LH concentrations, lownormal FSH levels, and increased LH:FSH ratios are typical, but more so in lean than in obese women with PCOS. In the past, an increased LH:FSH ratio (e.g., >2:1) has been regarded as a marker of PCOS, but the ratio varies with the assays used to measure gonadotropin concentrations, and the prevalence of obesity is high among women with PCOS.7,60, 307,308 and 309 Consequently, gonadotropin levels or ratios are not a reliable diagnostic criterion; they neither make, nor exclude, the diagnosis.
Insulin Resistance
Insulin resistance and hyperinsulinemia are common but not universal features of women with PCOS, no matter what method is used to assess insulin sensitivity.92,94,310 The overall prevalence of insulin resistance among women with PCOS is between 50% and 75%, and greater in obese than in lean women with PCOS.
Most women with PCOS and insulin resistance are young and have ample pancreatic β-cell reserve. Consequently, they are able to generate a compensatory hyperinsulinemia, allowing them to maintain normal glucose homeostasis, at least in the fasting state.100 Although most therefore display an exaggerated insulin response to a glucose challenge, some also exhibit evidence of β-cell dysfunction,311,312 and 313 particularly those having a family history of type 2 diabetes mellitus.314
Whereas there is no debate that insulin resistance and hyperinsulinemia play an important role in the pathophysiology of PCOS, or that the prevalence of unrecognized diabetes is sufficiently high to warrant testing to exclude the diagnosis in women with PCOS, the practical importance of detecting insulin resistance and what tests, if any, should be performed for that purpose remain highly controversial.
The gold standard method for measuring insulin sensitivity, to which all other methods are compared, is the hyperinsulinemic euglycemic clamp.315 The technique involves a fixedrate intravenous infusion of insulin and a simultaneous intravenous glucose infusion, varying the rate as needed to establish a steady state plasma glucose level within the normal fasting range. The glucose infusion rate at steady state estimates the rate of glucose uptake in tissues at the defined plasma insulin concentration and is inversely proportional to the degree of insulin resistance; the lower the glucose infusion rate at steady state, the greater the degree of insulin resistance. Insulin sensitivity is defined as the ratio of the glucose disposal rate to the steady state insulin concentration (glucose disposal rate [mmol/kG] × min per mU/L × 100). The clamp technique and other methods involving intravenous infusions of glucose and/or insulin (e.g., the frequently sampled intravenous glucose tolerance test, the insulin sensitivity test, the insulin tolerance test, and the continuous infusion of glucose with model assessment) have been used extensively in clinical investigations of glucose and insulin dynamics. However, they have no real practical clinical application because they are time-consuming, invasive, costly, and require experienced personnel.
The complexities of clamp techniques and other methods requiring intravenous infusions and multiple blood samplings spurred efforts to find an uncomplicated and inexpensive quantitative method for evaluating insulin sensitivity. A number of fasting state (homeostatic) measures have been described, all based on the fasting glucose and insulin concentrations and using straightforward calculations.316 One common weakness that all such methods have is that they assume a linear relationship between glucose and insulin that is, in fact, parabolic.
The fasting serum insulin concentration is easy to obtain and requires no calculations;317 in euglycemic White women with PCOS, values greater than 20-30 mU/mL suggest insulin resistance. The fasting glucose/insulin ratio has been used widely as an index of insulin sensitivity in women with PCOS; a ratio less than 4.5 has reasonable sensitivity and speci-ficity for insulin resistance.94 The homeostatic model assessment of insulin resistance (HOMA-IR) is another measure of insulin sensitivity commonly used in larger epidemiologic studies. The HOMA-IR is calculated by dividing the product of the fasting glucose (mg/dL) and insulin (mU/mL) concentrations by a constant: [glucose (mg/dL)][insulin (mU/mL)]/405, or [glucose (mmol/L)][insulin (mU/mL)]/22.5.318,319 The HOMA-IR value correlates relatively well with results from clamp studies,320,321 and unlike the fasting insulin concentration and the glucose/insulin ratio, compensates for fasting hyperglycemia;
values greater than 3.2-3.9 generally indicate insulin resistance.43,95,322 The quantitative insulin sensitivity check index (QUICKI) is yet another method for assessing insulin sensitivity in clinical investigations. Like the HOMA-IR, QUICKI can be applied in both euglycemic and hyperglycemic patients.323 The QUICKI value is the inverse of the sum of the fasting glucose and insulin concentrations, expressed logarithmically: (1/[log(Glucose)+log(Insulin)]); values greater than 0.33 indicate insulin resistance.43,324 Still other methods use a weighted combination of the fasting insulin and triglyceride concentrations and BMI to estimate insulin sensitivity.315 All of the calculated indices have limitations, primarily the lack of a standardized insulin assay. As the sheer number of different measures of insulin resistance demonstrates, there is no uniformly accepted test for measuring insulin resistance in a clinical setting. Consequently, routine screening for insulin resistance is not recommended.
values greater than 3.2-3.9 generally indicate insulin resistance.43,95,322 The quantitative insulin sensitivity check index (QUICKI) is yet another method for assessing insulin sensitivity in clinical investigations. Like the HOMA-IR, QUICKI can be applied in both euglycemic and hyperglycemic patients.323 The QUICKI value is the inverse of the sum of the fasting glucose and insulin concentrations, expressed logarithmically: (1/[log(Glucose)+log(Insulin)]); values greater than 0.33 indicate insulin resistance.43,324 Still other methods use a weighted combination of the fasting insulin and triglyceride concentrations and BMI to estimate insulin sensitivity.315 All of the calculated indices have limitations, primarily the lack of a standardized insulin assay. As the sheer number of different measures of insulin resistance demonstrates, there is no uniformly accepted test for measuring insulin resistance in a clinical setting. Consequently, routine screening for insulin resistance is not recommended.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


