INTRODUCTION
Evaluation and management of a patient with amenorrhea is common in gynecology. Specifically, the prevalence of pathologic amenorrhea ranges from 3 to 4 percent in reproductive-aged populations (Bachmann, 1982; Pettersson, 1973). Amenorrhea has classically been defined as primary (no prior menses) or secondary (cessation of menses). Although this distinction does suggest a relative likelihood of finding a particular diagnosis, the approach to diagnosis and treatment is similar for either presentation (Tables 16-1 and 16-2). Of course, amenorrhea is a normal state prior to puberty, during pregnancy and lactation, with certain hormonal medications such as continuous administration of combination oral contraceptives (COCs), and following menopause. Evaluation is considered for an adolescent: (1) who by age 13 has not menstruated or shown other evidence of pubertal development, or (2) who has reached other pubertal milestones but has not menstruated by age 15 or within 3 years of thelarche (American College of Obstetrician and Gynecologists, 2009). Secondary amenorrhea for 3 months or oligomenorrhea involving fewer than nine cycles a year is also investigated (American Society for Reproductive Medicine, 2008). In some circumstances, testing reasonably may be initiated despite the absence of these strict criteria. Examples include a patient with the stigmata of Turner syndrome, obvious virilization, or a history of uterine curettage. An evaluation for delayed puberty is also considered before the ages listed above if the patient or her parents are concerned.
| Presentation | Frequency (%) |
| Hypergonadotropic hypogonadism 45,X and variants 46,XX 46,XY | 43 27 14 2 |
| Eugonadism Müllerian agenesis Vaginal septum Imperforate hymen Androgen insensitivity syndrome Polycystic ovarian syndrome Congenital adrenal hyperplasia Cushing and thyroid disease | 30 15 3 1 1 7 1 2 |
| Low FSH without breast development Constitutional delay GnRH deficiency Other CNS disease Pituitary disease Eating disorders, stress, excess exercise | 27 14 5 1 5 2 |
| Etiology | Frequency (%) |
| Low or normal FSH level: various Eating disorders, stress, excess exercise Nonspecific hypothalamic Chronic anovulation (PCOS) Hypothyroidism Cushing syndrome Pituitary tumor/empty sella Sheehan syndrome | 67.5 15.5 18 28 1.5 1 2 1.5 |
| High FSH level: gonadal failure 46,XX Abnormal karyotype | 10.5 10 0.5 |
| High prolactin level | 13 |
| Anatomic Asherman syndrome | 7 7 |
| Hyperandrogenic states Nonclassic CAH Ovarian tumor Undiagnosed | 2 0.5 1 0.5 |
NORMAL MENSTRUAL CYCLE
A differential diagnosis for amenorrhea can be constructed based on requirements for normal menses. Ovarian function in a normal menstrual cycle is divided into the follicular phase (preovulatory), ovulation, and luteal phase (postovulatory). Endometrial characteristics are partitioned into the proliferative phase (preovulatory) and secretory phase (postovulatory). Generation of a cyclic, controlled pattern of uterine bleeding requires precise temporal and quantitative regulation of several reproductive hormones (Chap. 15).
First, the hypothalamic-pituitary-ovarian axis must be functional. The hypothalamus releases pulses of gonadotropin-releasing hormone (GnRH) into the hypophyseal portal circulation at defined frequencies and amplitude. GnRH stimulates the synthesis and secretion of the gonadotropins luteinizing hormone (LH) and follicle-stimulating hormone (FSH) by the gonadotrope cells of the anterior pituitary gland. These gonadotropins enter the peripheral circulation and act on the ovary to stimulate both follicular development and ovarian hormone production. These ovarian hormones include estrogens, progesterones, and androgens. Gonadal steroids are typically inhibitory at both the pituitary and the hypothalamus. Development of a mature follicle, however, creates a rapid rise in estrogen levels, which instead act positively to generate an LH surge. This surge is essential for ovulation.
Following ovulation, LH stimulates luteinization of the granulosa and theca cells, which had surrounded the mature oocyte, to form the corpus luteum. The corpus luteum continues to produce estrogen but also secretes high levels of progesterone. The thickened, proliferative endometrial lining produced by high circulating estrogen levels during the follicular phase is now converted to a secretory pattern by this luteal progesterone. If pregnancy occurs, the corpus luteum is “rescued” by human chorionic gonadotropin (hCG) secreted from early placental trophoblast. hCG is similar structurally to LH, shares the same receptor as LH, and assumes the role of corpus luteum support during early pregnancy. If pregnancy does not occur, then progesterone and estrogen secretion ceases, the corpus luteum regresses, and the endometrium sloughs. The pattern of this “progesterone withdrawal bleed” varies in duration and amount among women but is relatively constant across cycles for a given individual.
Amenorrhea may follow disruption of this choreographed communication. However, even with normal hormonal cycling, altered anatomy may prevent menses. The endometrium must be able to respond normally to hormonal stimulation, and the cervix, vagina, and introitus must be present and patent.
CLASSIFICATION SYSTEM
Numerous classification systems for the diagnosis of amenorrhea have been developed, and all have their strengths and weaknesses. One useful scheme is outlined in Table 16-3. This system divides causes of amenorrhea into anatomic versus hormonal etiologies, with further division into congenital versus acquired disorders.
As described above, normal menses require adequate ovarian production of steroid hormones. Decreased ovarian function (hypogonadism) may result either from a lack of stimulation by the gonadotropins (hypogonadotropic hypogonadism) or from primary failure of the ovary (hypergonadotropic hypogonadism) (Table 16-4). Several disorders are associated with relatively normal LH and FSH levels (eugonadotropic), however, appropriate cyclicity is lost.
ANATOMIC DISORDERS
Anatomic abnormalities causing amenorrhea can broadly be viewed as either inherited or acquired disorders of the outflow tract (uterus, cervix, vagina, and introitus). Of these two, an inherited cause is frequent in adolescents, and pelvic anatomy is abnormal in approximately 15 percent of women with primary amenorrhea (American Society for Reproductive Medicine, 2008). Figure 16-1 depicts the range of anatomic defects that may present with amenorrhea. These are additionally discussed in Chapter 18.
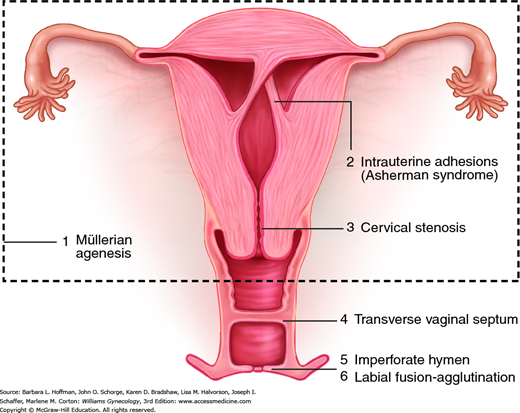
Amenorrhea is associated with imperforate hymen (1 in 2000 women), a complete transverse vaginal septum (1 in 70,000 women), or isolated vaginal atresia (Banerjee, 1998; Parazzini, 1990). Also, although structurally normal, labia in some girls may be severely agglutinated and can lead to obstruction. Most with agglutination are treated early with topical estrogen and/or manual separation, and outflow obstruction is thereby avoided.
Patients with outflow obstruction have a 46,XX karyotype, female secondary sexual characteristics, and normal ovarian function. Thus, the amount of uterine bleeding is normal, but its normal path for egress is obstructed or absent. Patients may note moliminal symptoms, such as breast tenderness, food cravings, and mood changes, which are attributable to elevated progesterone levels. With inherited or acquired outflow obstruction, accumulation of blood behind the blockage frequently results in cyclic abdominal pain. Intrauterine trapping of fluid (hydrometra), pus (pyometra), or blood (hematometra) creates a soft, enlarged uterus. Similarly, the terms hydrocolpos, pyocolpos, and hematocolpos describe distention of the vagina and are seen with distal obstructions. Moreover, in women with outflow blockage, an increase in retrograde menstruation may lead to endometriosis development.
During embryonic development, the müllerian ducts give rise to the upper vagina, cervix, uterine corpus, and fallopian tubes. Agenesis during these ducts’ development may be partial or complete. Accordingly, amenorrhea may result from outflow obstruction or from a lack of endometrium in cases involving uterine agenesis. In complete müllerian agenesis, often called Mayer-Rokitansky-Kuster-Hauser syndrome, patients fail to develop any müllerian structures, and examination reveals only a vaginal dimple (Chap. 18). It ranks second only to gonadal dysgenesis as a cause of primary amenorrhea (Aittomaki, 2001; Reindollar, 1981).
Similar to patients with lower outflow tract obstruction, patients with müllerian anomalies have a 46,XX karyotype and normal ovarian function. Research has begun to identify candidate gene mutations that may contribute to this disorder, but details are lacking. Importantly, complete müllerian agenesis may be confused with complete androgen insensitivity syndrome. In the latter condition, the patient has a 46,XY karyotype and functioning testes. However, underlying androgen receptor mutations prevent normal testosterone binding, normal male ductal system development, and virilization. These two syndromes are compared in Table 16-5 and discussed further in Chapter 18.
| Presentation | Müllerian Agenesis | Androgen Insensitivity |
| Inheritance pattern Karyotype Breast development Axillary and pubic hair Uterus Gonad Testosterone Associated anomalies | Sporadic 46,XX Yes Yes No Ovary Female levels Yes | X-linked recessive 46,XY Yes No No Testis Male levels No |
Other abnormalities of the uterus that cause amenorrhea include cervical stenosis and extensive intrauterine adhesions. With stenosis, postoperative scarring and cervical os narrowing may follow dilatation and curettage (D & C), cervical conization, loop electrosurgical excision procedures, infection, and neoplasia. Severe atrophic or radiation changes are other sources. Stenosis most commonly involves the internal os, and symptoms in menstruating women include amenorrhea or abnormal bleeding, dysmenorrhea, and infertility. Management seeks to reopen the os and is discussed in Chapter 4.
Also known as uterine synechiae and, when symptomatic, as Asherman syndrome, the spectrum of scarring includes filmy adhesions, dense bands, or complete obliteration of the uterine cavity (Fig. 16-2). Normally, the endometrium is divided into a functional layer, which lines the endometrial cavity, and a basal layer, which regenerates the functional layer with each menstrual cycle. Destruction of the basal endometrium prevents endometrial thickening in response to ovarian steroids. Thus, no tissue is produced or subsequently sloughed when steroid hormone levels fall at the end of the luteal phase.
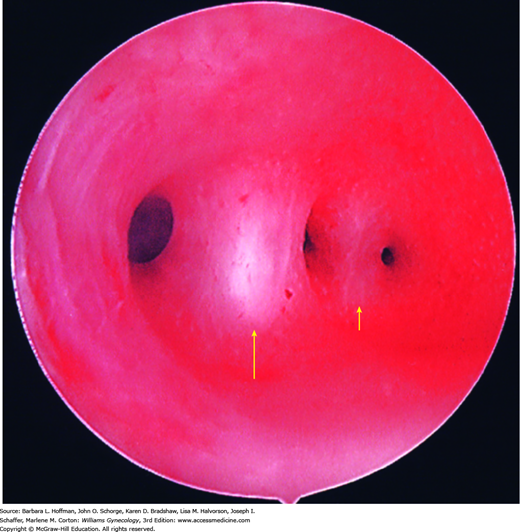
Endometrial damage may follow vigorous curettage, usually in association with postpartum hemorrhage, miscarriage, or elective abortion complicated by infection. In a series of 1856 women with Asherman syndrome, 88 percent followed postabortal or postpartum uterine curettage (Schenker, 1982). Damage may also result from other uterine surgery, including metroplasty, myomectomy, or cesarean delivery, or from infection related to an intrauterine device. Although rare in the United States, tuberculous endometritis is a relatively common cause of Asherman syndrome in developing countries (Sharma, 2009). Of course, Asherman syndrome may also be an intentional outcome following uterine ablation for heavy menstrual bleeding.
Depending on the degree of scarring, patients may describe amenorrhea; in less severe cases, hypomenorrhea; or recurrent pregnancy loss due to inadequate placentation (March, 2011). In their evaluation of 292 women with intrauterine adhesions, Schenker and Margalioth (1982) noted delivery of term pregnancies in only 30 percent of 165 pregnancies. The remaining pregnancies either were spontaneously aborted (40 percent) or delivered prematurely.
If intrauterine adhesions are suspected, radiologic evaluation of the uterine cavity is performed. If information regarding tubal patency is needed, then hysterosalpingography (HSG) offers information regarding both uterine cavity and fallopian tube patency. Otherwise, saline infusion sonography (SIS) provides an excellent alternative. Intrauterine adhesions characteristically appear as irregular, angulated filling defects within the cavity (Fig. 19-6 and Fig. 2-23). At times, uterine polyps, leiomyomas, air bubbles, and blood clots may masquerade as adhesions. Definitive diagnosis requires hysteroscopy. To improve fertility rates or to relieve symptomatic hematometra, hysteroscopic lysis of adhesions is the preferred surgical treatment. The procedure is described in Section 44-19, and fertility advantages are discussed in Chapter 20.
HYPERGONADOTROPIC HYPOGONADISM
The term hypergonadotropic hypogonadism describes any process in which: (1) ovarian function is decreased or absent (hypogonadism) and (2) due to absent negative sex-steroid feedback, the gonadotropins, LH and FSH, have increased serum levels (hypergonadotropic). This category of disorders implies primary dysfunction within the ovary rather than hypothalamic or pituitary dysfunction (Table 16-6). This process can also be termed premature menopause or premature ovarian failure (POF), with a current trend toward the term premature ovarian insufficiency or primary ovarian insufficiency (POI). The term ovarian insufficiency conveys the fact that ovarian function may fluctuate before complete oocyte depletion and may lead to occasional transient menses resumption or even pregnancy (Bidet, 2011). For our purposes, the term premature ovarian failure, implying permanent cessation of menses, will be used.
POF is defined as loss of oocytes and the surrounding support cells prior to age 40 years. The diagnosis is determined by two serum FSH levels that measure greater than a threshold range of 30 to 40 mIU/mL and are obtained at least 1 month apart. This definition distinguishes POF from the physiologic loss of ovarian function, which occurs with normal menopause. The incidence of POF has been estimated at 1 in 1000 women younger than 30 years and at 1 in 100 women younger than 40 (Coulam, 1986).
Careful evaluation is mandatory as the diagnosis and effective treatment may have significant implications for the patient’s psychologic, cardiovascular, bone, and sexual health. The finding of a genetic disorder may also require evaluation of family members. Nevertheless, in most cases, an etiology for POF is not determined (American College of Obstetricians and Gynecologists, 2014).
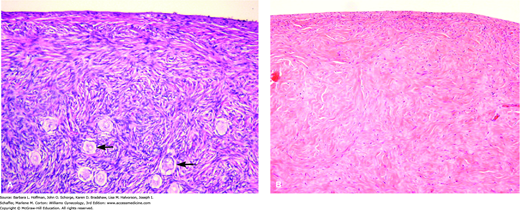
Gonadal dysgenesis is the most frequent cause of POF. In this disorder, a normal complement of germ cells is present in the early fetal ovary. However, oocytes undergo accelerated atresia, and the ovary is replaced by a fibrous streak—termed a streak gonad (Figs. 16-3 and 16-4)(Simpson, 1975; Singh, 1966). Individuals with gonadal dysgenesis may present with various clinical features and can be divided into two broad groups based on whether their karyotype is normal or abnormal (Schlessinger, 2002). These are all discussed further in Chapter 18.
FIGURE 16-3
Photomicrographs of histologic samples. A. Normal premenopausal ovarian cortex with multiple primordial follicles (arrows). (Used with permission from Dr. Kelley Carrick.) B. Ovary from a woman with gonadal dysgenesis. Streak ovary showing ovarian-type stroma with no primordial follicles (Used with permission from Dr. Raheela Ashfaq.)
Those with normal karyotype (46,XX or 46,XY) are described as having “pure” gonadal dysgenesis. Patients with a 46,XY genotype and gonadal dysgenesis (Swyer syndrome) are phenotypically female due to absent testosterone and absent antimüllerian hormone (AMH) secretion by the dysgenetic testes. The etiology of the gonadal failure in both genetically male and female patients is poorly understood but is likely due to single gene defects or destruction of gonadal tissue in utero, perhaps by infection or toxins (Hutson, 2014).
Those with abnormal karyotype include Turner syndrome (45,X) and chromosomal mosaics such as 45,X/46,XX or 45,X/46,XY. In general, approximately 90 percent of individuals with gonadal dysgenesis from a loss of X genetic material never menstruate. The remaining 10 percent have sufficient residual follicles to experience menses and rarely may achieve pregnancy. However, the menstrual and reproductive lives of such individuals are invariably brief (Kaneko, 1990; Simpson, 1975; Tho, 1981).
As noted, in some cases of gonadal dysgenesis, a Y chromosome is present. If Y chromosomal material is found, the streak gonads are removed because nearly 25 percent of these patients will develop a malignant germ cell tumor (Chap. 36) (Manuel, 1976). Thus, chromosomal analysis is performed in all cases of amenorrhea associated with POF, particularly before age 30. The presence of a Y chromosome cannot be determined clinically, as only a few patients will demonstrate signs of androgen excess.
In addition to chromosomal abnormalities, patients may experience POF due to single gene mutations (Cordts, 2011; Goswami, 2005). First, a significant relationship is noted between fragile X syndrome and POF (American College of Obstetricians and Gynecologists, 2010). This syndrome is caused by a triple repeat sequence mutation in the X-linked FMR1 (fragile X mental retardation) gene. This gene is unstable, and its size can expand during parent-to-child transmission. The fully expanded mutation (>200 CGG repeats) becomes hypermethylated, resulting in silencing of gene expression. As such, this fully expanded mutation is the most common known inherited genetic cause of mental retardation and of autism. Males with the so-called premutation (50 to 200 CGG repeats) are at risk for fragile-X associated tremor/ataxia syndrome (FXTAS). Females with the premutation have a 13- to 26-percent risk of developing POF, although the mechanism is unclear. An estimated 0.8 to 7.5 percent of sporadic POF and 13 percent of familial POF cases are due to premutations in this gene. The prevalence of premutations in women approximates 1 in 129 to 300 (Wittenberger, 2007).
Less common gene defects are CYP17 mutations. These decrease 17α-hydroxylase and 17,20-lyase activity and thereby prevent production of cortisol, androgens, and estrogens (Fig. 15-5). Affected patients have sexual infantilism and primary amenorrhea due to absent estrogen secretion. Sexual infantilism describes patients with a lack of breast development, absent pubic and axillary hair, and a small uterus. Mutations in CYP17 also increase adrenocorticotropin hormone (ACTH) release, thereby stimulating mineralocorticoid secretion. This, in turn, leads to hypokalemia and hypertension (Goldsmith, 1967).
Mutations in genes that encode LH and FSH receptors have also been reported. These defects prevent normal responses to circulating gonadotropins, a condition termed resistant ovary syndrome (Aittomaki, 1995; Latronico, 2013). Identification of other single gene mutations that can cause POF is an area of active investigation. The list of implicated genes now includes those that encode both estrogen receptors (ERα and ERβ), extracellular signaling proteins (specifically, BMP15), and transcription factors FOXL2, FOX03, and SF-1 (Cordts, 2011; Goswami, 2005). The importance of these factors in maintaining ovarian function is providing further insights into normal ovarian physiology and may lead to new infertility treatments and contraceptive options.
Although frequently cited, galactosemia is a rare cause of POF. Classic galactosemia affects 1 in 30,000 to 60,000 live births. Inherited as an autosomal recessive disorder, this condition leads to abnormal galactose metabolism due to a deficiency of galactose-1-phosphate uridyl transferase, encoded by the GALT gene (Rubio-Gozalbo, 2010). Galactose metabolites are believed to have a direct toxic effect on many cell types, including germ cells. Potential complications include neonatal death, ataxic neurologic disease, cognitive disabilities, and cataracts. POF will develop in almost 85 percent of females if left untreated. Treatment is lifelong dietary restriction of galactose, which is present in milk-based foods. Galactosemia is frequently diagnosed during newborn screening programs or during pediatric evaluation of impaired growth and development and long before a patient would present to a gynecologist (Kaufman, 1981; Levy, 1984).
Hypergonadotropic hypogonadism can be acquired from infection, environmental exposures, autoimmune disease, or medical treatments. Of these, infectious causes of POF are relatively rare and poorly understood, with mumps oophoritis being the most frequently reported (Morrison, 1975). Various environmental toxins have a clear detrimental effect on follicular health. These include cigarette smoking, heavy metals, solvents, pesticides, and industrial chemicals (Jick, 1977; Mlynarcikova, 2005; Sharara, 1998).
Autoimmune disorders account for an estimated 40 percent of POF cases (Hoek, 1997; LaBarbera, 1988). Ovarian failure may be one component of autoimmune pituitary polyglandular failure and accompanied by hypothyroidism and adrenal insufficiency, or it may follow other autoimmune disorders such as systemic lupus erythematosus. POF has also been associated with myasthenia gravis, idiopathic thrombocytopenic purpura, rheumatoid arthritis, vitiligo, and autoimmune hemolytic anemia (de Moraes, 1972; Jones, 1969; Kim, 1974). Although several antiovarian antibodies have been characterized, there is currently no validated serum antibody marker to assist in the diagnosis of autoimmune POF (American Society for Reproductive Medicine, 2008).
Iatrogenic ovarian failure is relatively common. This group includes patients who have undergone surgical removal of the ovaries or cystectomy due to recurrent ovarian cysts, endometriosis, or severe pelvic inflammatory disease. Alternatively, a woman may experience amenorrhea following pelvic radiation for cancer or following chemotherapy for treatment of malignancies or severe autoimmune disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


