Cardiac Disorders in Pregnancy
Sreedhar Gaddipati
Nan H. Troiano
The physiologic changes associated with pregnancy are significant, yet the healthy gravida is able to accommodate these changes without difficulty. In the presence of a cardiac disorder, however, the pregnant woman may face significant risks for morbidity or mortality.
Cardiac disorders have been reported to occur in 0.5 to 4.0 percent of all pregnancies and represent one of the most important nonobstetric causes of maternal death.1,2,3,4,5 In the United States, the maternal death rate has risen from 7.2 deaths per 100,000 live births in 1987 to 13 deaths per 100,000 live births in 2004.6 While the leading causes of maternal death in the U.S. are embolism, hemorrhage, hypertension, and infection, cardiac disease, specifically cardiomyopathy, has increased.7 Another factor is the increasing population of women with underlying congenital heart disease (CHD). It is estimated that the adult population with CHD is increasing by 5 percent per year.8 As the increased number of women with successfully managed CHD reach childbearing age, the need for appropriate collaborative care across the specialty practice of women’s health is crucial.
This chapter describes the classification of cardiac disorders in pregnancy, presents information regarding specific cardiac lesions, and illustrates how selected lesions impact the woman during pregnancy. The necessity for collaborative management throughout pregnancy is emphasized, and data from clinical case studies are presented to illustrate significant concepts.
Classification
Cardiac disease during pregnancy may generally be categorized as congenital, acquired, or ischemic in nature. The utility of a classification system is to facilitate prediction of risk with respect to how pregnancy will be tolerated by both the pregnant woman with a cardiac disorder and her fetus, and to enhance the quality of patient counseling. It is preferable that women with a known cardiac disorder be counseled prior to conception so that they have a more thorough understanding of their specific risk for morbidity and mortality should they become pregnant. However, since 49 percent of pregnancies are unplanned, and the presence of a cardiac disorder may not be known, or may be diagnosed after the pregnancy has progressed, all obstetric health care providers should be familiar with signs and symptoms of cardiac dysfunction and the risks associated with pregnancy.9
The risk of perinatal morbidity and mortality for the pregnant woman with preexisting cardiac disease is dependent on three factors: (1) the specific cardiac lesion, (2) the functional abnormality produced by the lesion, and (3) the development of complications such as hemorrhage, infection, and pregnancy-induced hypertension.10 A classification system that identifies the risk of mortality associated with specific cardiac lesions during pregnancy has been described and is presented in Table 8-1.11,12 In addition, risk factors predictive of congestive heart failure, stroke, or arrhythmias during the pregnancy of a woman with cardiac disease have been described and are presented in Tables 8-2A and 8-2B.13,14
Functional ability also influences pregnancy outcome. Prior to 1973, the Criteria Committee of the New York Heart Association (NYHA) recommended a classification system of cardiac disease based on clinical function. This system is currently utilized as part of a thorough assessment of the pregnant woman with cardiac disease. A description of the NYHA functional classification system is found in Table 8-3. Patients classified as NYHA class I or II prior to pregnancy generally do well during pregnancy. Those with functional classification III or IV have a significantly increased risk of morbidity and mortality during pregnancy. Although this system is useful in caring for the pregnant patient with cardiac disease, it has distinct limitations with respect to its ability to predict successful pregnancy outcome. As many as 40 percent of women who develop congestive heart failure
and pulmonary edema during pregnancy are functional class I prior to pregnancy. 15 Therefore, it is imperative that pregnant women with cardiac disorders be assessed at each prenatal visit, hospital presentation, or admission, and reclassified incorporating any adverse change in functional ability.
and pulmonary edema during pregnancy are functional class I prior to pregnancy. 15 Therefore, it is imperative that pregnant women with cardiac disorders be assessed at each prenatal visit, hospital presentation, or admission, and reclassified incorporating any adverse change in functional ability.
Table 8.1 Mortality Risk Associated with Pregnancy and Cardiac Disorders | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
Table 8.2A Predictors of Congestive Heart Failure, Stroke, or Arrhythmia | |
|---|---|
|
Table 8.2B Risk of Congestive Heart Failure, Stroke, or Arrhythmia During Pregnancy by Number of Risk Factors | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Table 8.3 NYHA Functional Classification System | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Congenital Cardiac Disorders
Incidence
The incidence of congenital heart disease (CHD) is 9 per 1,000 liveborn infants. The incidence is higher if infants are included who have bicuspid aortic valves (incidence of 13.7 per 1,000 adults), or if the approximately 5 percent of all infants who are born with small ventriculoseptal defects that spontaneously close are included.16,17 The frequency of congenital as opposed to acquired cardiac disease has increased.8 Two factors have contributed to this change.18,19 First, advances in neonatal intensive care and pediatric cardiac surgery have allowed children with CHD to reach reproductive age. Second, the incidence of rheumatic fever in the U.S. as well as worldwide has decreased significantly over time. The ratio of rheumatic heart disease to CHD seen during pregnancy decreased from approximately 20:1 in the early 1950s to 3:1 by the late 1970s. Currently this ratio has approached unity, and in some populations the ratio has reversed.20
Etiology
Based on earlier studies regarding the risk of recurrence and transmission, a hypothesis of multifactorial etiology was proposed for CHD, which attributed these defects to interactions between genetic predisposition and environmental influences.21 However, more recent
studies suggest separate genetic and environmental causes.19 As cytogenetic testing techniques have advanced, high-resolution karyotypic analysis has more precisely revealed chromosome deletions, duplication, and translocations, some of which are very subtle. Analysis of the telomeric and subtelomeric regions has led to discovery of such variants; one of the most recognizable is 22q11 deletion. This deletion has been estimated to occur in 1 per 5,950 live births, and has been associated with DiGeorge syndrome and velocardiofacial syndrome. Similarly, Williams-Beuren syndrome has been found, as a result of analysis by both fluorescence in situ hybridization (FISH) and DNA mutation, to have a microdeletion at chromosome 7q11.23. Linkage analysis and polymerase chain reaction (PCR) are techniques that have helped identify single genes that may be linked to both syndromic (e.g., Noonan syndrome, Holt-Oram syndrome) and nonsyndromic CHD.
studies suggest separate genetic and environmental causes.19 As cytogenetic testing techniques have advanced, high-resolution karyotypic analysis has more precisely revealed chromosome deletions, duplication, and translocations, some of which are very subtle. Analysis of the telomeric and subtelomeric regions has led to discovery of such variants; one of the most recognizable is 22q11 deletion. This deletion has been estimated to occur in 1 per 5,950 live births, and has been associated with DiGeorge syndrome and velocardiofacial syndrome. Similarly, Williams-Beuren syndrome has been found, as a result of analysis by both fluorescence in situ hybridization (FISH) and DNA mutation, to have a microdeletion at chromosome 7q11.23. Linkage analysis and polymerase chain reaction (PCR) are techniques that have helped identify single genes that may be linked to both syndromic (e.g., Noonan syndrome, Holt-Oram syndrome) and nonsyndromic CHD.
Similarly, CHD has been associated with non-inheritable risk factors. Wilson, et al., estimated that up to 30 percent of these factors may be modifiable, thus suggesting that the incidence may potentially be decreased.22,23 Examples of maternal diseases that increase the risk of fetal congenital cardiac disorders include diabetes mellitus, lupus erythematosus, and phenylketonuria. Systemic lupus erythematosus, while not associated with structural disease, has been associated with heart block via ssA and ssB antibodies, which damage the conduction pathways within the fetal heart. Another example is maternal exposure to environmental factors. This category includes viral illness, drug exposure, and factors within the home or workplace. Febrile illness, influenza, and rubella viral infection have been associated with CHD. Similarly, exposure to drugs such as vitamin A, other retinoids, and anticonvulsants has been linked to CHD. Lithium, once linked to increased incidence of Ebstein’s anomaly, is no longer thought to be a cardiac teratogen. While there are no data to support a causative association between caffeine and CHD, alcohol ingestion remains of concern.23
It is clear that further research is needed to identify both genetic and environmental causes in order to provide effective prenatal counseling, and to potentially modify risk.
Of equal concern in pregnant women with CHD is the risk of fetal congenital cardiac anomalies. The risk of fetal cardiac anomalies is approximately 5 percent, although the actual incidence may be higher in women whose congenital lesion involves ventricular outflow obstruction. During the antepartum period, serial ultrasonography should be performed in order to assess the fetus for appropriate interval growth. Fetal echocardiography is indicated for prenatal diagnosis of congenital cardiac defects. Of special interest is that affected fetuses appear to be concordant for the maternal lesion in approximately 50 percent of cases.12 Figure 8-1 depicts an echocardiographic image of a fetus at 32 weeks and 5 days gestational age in a pregnant woman with a ventricular septal defect (VSD). A similar VSD is demonstrated in this fetus.
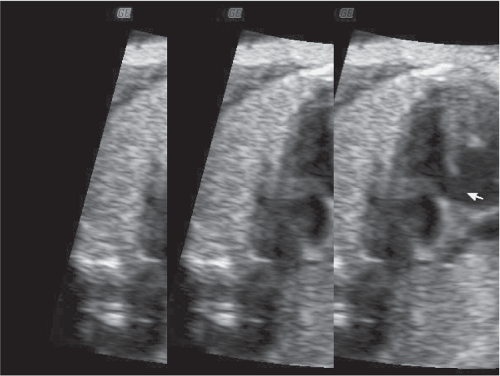 Figure 8-1 Echocardiographic image of a fetus at 32 weeks and 5 days’ gestation in a pregnant woman with a VSD. A similar VSD is demonstrated in this fetus. (The defect is noted by the arrow.) |
Left-to-Right Shunts
Atrial Septal Defect
Atrial septal defect (ASD) is one of the most common congenital cardiac lesions found during pregnancy, and
is the third most common in adults.1 In a study of a series of 113 women with a known ASD, one death was noted in 219 pregnancies.24 Three important potential complications seen with ASDs are arrhythmias, paradoxical embolism, and congestive heart failure (CHF). Figures 8-2A and 8-2B depict cardiac abnormalities documented following the death of a pregnant woman from complications of an ASD. Figure 8-2A demonstrates the right atrium to be significantly thin, almost transparent. A two-part ASD is visible with a trabeculated area. The ASD is significantly large, measuring 4 centimeters. Figure 8-2B reveals the presence of a thickened, myxomatous mitral valve, in addition to the ASD. Myxomatous mitral valve disease is a disorder characterized by enlarged, thickened, floppy, gelatinous leaflets, and elongated chordae tendineae. The myxomatous valve as a whole is often moderately to severely regurgitant, which means that blood may leak back through the valve into the left atrium. A summary of the autopsy findings in this case included: a fenestrated ASD measuring 4 centimeters, significant myxomatous changes of the mitral valve consistent with severe mitral valve prolapse, dilation of the right heart chambers, and absence of evidence of a pulmonary embolism. The likely cause of death was noted to be cardiac arrhythmia.
is the third most common in adults.1 In a study of a series of 113 women with a known ASD, one death was noted in 219 pregnancies.24 Three important potential complications seen with ASDs are arrhythmias, paradoxical embolism, and congestive heart failure (CHF). Figures 8-2A and 8-2B depict cardiac abnormalities documented following the death of a pregnant woman from complications of an ASD. Figure 8-2A demonstrates the right atrium to be significantly thin, almost transparent. A two-part ASD is visible with a trabeculated area. The ASD is significantly large, measuring 4 centimeters. Figure 8-2B reveals the presence of a thickened, myxomatous mitral valve, in addition to the ASD. Myxomatous mitral valve disease is a disorder characterized by enlarged, thickened, floppy, gelatinous leaflets, and elongated chordae tendineae. The myxomatous valve as a whole is often moderately to severely regurgitant, which means that blood may leak back through the valve into the left atrium. A summary of the autopsy findings in this case included: a fenestrated ASD measuring 4 centimeters, significant myxomatous changes of the mitral valve consistent with severe mitral valve prolapse, dilation of the right heart chambers, and absence of evidence of a pulmonary embolism. The likely cause of death was noted to be cardiac arrhythmia.
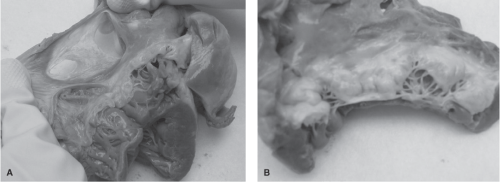 Figure 8-2 (A) The right atrium is significantly thin, almost transparent. A two-part ASD is visible with a trabeculated area. The ASD is significantly large, measuring 4 centimeters. (B) The presence of a thickened, myxomatous mitral valve, in addition to the ASD, is visible in this image. |
The presence of a large ASD allows for an increased risk for paradoxical emboli, and consideration should be paid to venous thromboembolic (VTE) prophylaxis during labor and the postpartum period, either by application of venous compression devices or administration of prophylactic heparin. Hypervolemia associated with pregnancy results in an increased left-to-right shunt of blood through the ASD, which inflicts a significant burden on the right ventricle.25 Although the subsequent additional right preload is tolerated well by most patients, CHF and death have been reported.26 ASD is usually characterized by normal to low pulmonary artery pressures. Therefore, the development of pulmonary hypertension is uncommon. Although the majority of pregnant women with an ASD tolerate pregnancy, labor, and delivery well, significant morbidity and mortality may occur as a result of complications from an ASD during pregnancy.
Ventricular Septal Defect
Ventricular septal defect (VSD) may occur as a single lesion or in combination with other congenital cardiac anomalies, such as tetralogy of Fallot or transposition of the great vessels. Many VSDs are diagnosed by ultrasound in utero and corrected soon after birth. The majority of VSDs are diagnosed and repaired before the woman reaches childbearing age. In women with an uncorrected VSD, the size of the defect is the most important variable when one evaluates the risk of development of complications during pregnancy. Risk is associated primarily with fluid overload. In patients with a small defect, labor, delivery, and postpartum are usually tolerated without difficulty. Larger defects are associated more commonly with CHF or the development of pulmonary hypertension, as filling pressures increase within chambers on the right side of the heart. A large VSD may also be associated with aortic regurgitation, which can increase the risk of CHF. As with ASD, there are more consequences from embolus formation; thus, prophylaxis should be considered. A significant risk associated with a large VSD is development of right-to-left shunting of blood, thereby
bypassing the oxygenation process and exchange of gases that normally occur in the lungs. This condition, referred to as shunting, causes a significant percentage of blood pumped from the left ventricle to leave the heart without oxygen. Although cardiac output may remain normal, oxygen transport is severely impaired and can lead to multiple organ system dysfunction or failure, and maternal death.
bypassing the oxygenation process and exchange of gases that normally occur in the lungs. This condition, referred to as shunting, causes a significant percentage of blood pumped from the left ventricle to leave the heart without oxygen. Although cardiac output may remain normal, oxygen transport is severely impaired and can lead to multiple organ system dysfunction or failure, and maternal death.
Patent Ductus Arteriosus
Although a common congenital cardiac anomaly, patent ductus arteriosus (PDA) is generally detected and closed during the newborn period. Therefore, it is an unusual finding during pregnancy. Patients who present with a PDA during pregnancy usually tolerate the hemodynamic stress of pregnancy, labor, and delivery without difficulty. However, the high-pressure, high-flow, left-to-right shunt associated with a large uncorrected PDA can result in the development of pulmonary hypertension or CHF.25
Other Congenital Lesions
Tetralogy of Fallot
Tetralogy of Fallot refers to the complex of four cardiac lesions: ventricular septal defect, overriding aorta, pulmonary stenosis, and right ventricular hypertrophy. It is the most common cyanotic heart defect in individuals who survive to adulthood, and accounts for approximately 10 percent of all CHD.27 The major physiologic risk posed by tetralogy of Fallot is the potential shunting of blood past the lungs without being oxygenated. As much as 75 percent of the venous blood that returns to the heart may pass directly from the right ventricle into the aorta without becoming oxygenated. Most cases are surgically corrected in early childhood. The procedure involves reduction of the pulmonary stenosis, closure of the septal defect, and reconstruction of the flow pathway into the aorta. Although there are late sequelae of this surgery, long-term survival is 86 percent at 32 years.28,29 The majority of patients with corrected tetralogy of Fallot experience good outcomes in pregnancy. Meijer and colleagues reviewed the cases of 29 women with tetralogy of Fallot who underwent 63 pregnancies.30 Thirteen of these resulted in abortion, twelve of which were spontaneous. Of the remaining 50 successful pregnancies, statistics were available on 46 deliveries. Five delivered between 16 and 36 weeks, and the remaining were >37 weeks. Of the 26 women who had successful pregnancies, five experienced cardiac events in six pregnancies. Each of these patients experienced either ventricular tachycardia (VT) or supraventricular tachycardia (SVT), and two experienced right-sided cardiac failure.30 It should be noted that all of these women were NYHA class I prior to pregnancy, further emphasizing the importance of reassessing patients at each visit to determine if NYHA reclassification is necessary. Patients with an uncorrected VSD may experience worsening of the right-to-left shunt related to the decrease in systemic vascular resistance (SVR) that accompanies pregnancy. Poor prognosis has been associated with patients who have a prepregnancy hematocrit greater than 65 percent, a history of syncopal episodes, CHF, oxygen saturation less than 90 percent, high right-ventricular pressures, or cardiomegaly.1 Death may occur following surgical repair secondary to arrhythmias, sudden coronary death (6 to 9 percent risk after 30 years), CHF, and complications from subsequent surgery. Thus, while corrected tetralogy of Fallot is usually well tolerated in pregnancy, a good outcome should not be presumed. Cardiac consultation and evaluation should be included as part of prepregnancy counseling of women with corrected tetralogy of Fallot.
Coarctation of the Aorta
The most common site of coarctation is distal to the left subclavian artery.1 This condition is most commonly seen with a bicuspid aortic valve but also may be associated with aortic stenosis or regurgitation, thoracic aortopathy, aneurysm of the circle of Willis, and the cardiac and systemic results of long-standing arterial hypertension. 24 If left untreated, patients with coarctation of the aorta are at increased risk for the development of arterial hypertension, CHF, infectious aortitis or endocarditis, myocardial infarction, cerebrovascular accidents, aortic aneurysms, and dissection or rupture complicating aortopathy.25
Historically, pregnancy in women with coarctation of the aorta was discouraged. In a review of 200 pregnant women with coarctation of the aorta before 1940, Mendelson reported 14 maternal deaths and thus concluded that pregnancy, labor, and delivery posed significant risk to patients with this disorder.31 Consequently, contraception, therapeutic abortion, Cesarean section, and sterilization were among the therapeutic options recommended for women with this disorder. Similar recommendations were reported in the literature 20 years later.32 Deal and Wooley reported the outcomes of 28 women with uncorrected coarctation of the aorta who had 83 pregnancies.33 All were classified as NYHA class I or II prior to pregnancy. In this group of women, no maternal deaths or permanent cardiovascular complications occurred. More recently, Beauchesne and colleagues reported the pregnancy outcomes in a study that included 10 pregnant women with uncorrected coarctation of the aorta and 30 pregnant women who had successfully repaired coarctation of the aorta.34 Hypertension was noted in 30 percent of the total pregnancies. One maternal death occurred in the study population. That patient also had Turner syndrome and
underwent surgery at age 4 to repair coarctation of her aorta. Aortic dissection occurred during her subsequent pregnancy at 36 weeks gestation, from which complications developed that resulted in her death. Vriend and colleagues reported on the outcomes of 54 women from the Netherlands who experienced 126 pregnancies.35 Results included 98 live births; six were preterm and the gestational age at delivery ranged from 28 to 43 weeks. Twenty-eight spontaneous pregnancy losses were reported. Twenty-six (22 percent) of the pregnancies were complicated by hypertension.
underwent surgery at age 4 to repair coarctation of her aorta. Aortic dissection occurred during her subsequent pregnancy at 36 weeks gestation, from which complications developed that resulted in her death. Vriend and colleagues reported on the outcomes of 54 women from the Netherlands who experienced 126 pregnancies.35 Results included 98 live births; six were preterm and the gestational age at delivery ranged from 28 to 43 weeks. Twenty-eight spontaneous pregnancy losses were reported. Twenty-six (22 percent) of the pregnancies were complicated by hypertension.
Assessment of the aortic gradient may also be useful in predicting pregnancy outcome in patients with coarctation of the aorta. In general, aortic gradients across the site of coarctation that are less than 20 mmHg are associated with good maternal and fetal outcomes. Thus, with close monitoring and treatment of blood pressure, this lesion is usually associated with a good outcome during pregnancy.1,27 However, for those with additional manifestations of this defect, pregnancy may pose increased risk. For example, patients with an untreated aortic aneurysm or aneurysm in the circle of Willis may have a mortality rate as high as 15 percent. Thus, pregnancy termination for such patients may be considered.25
Eisenmenger Syndrome
Eisenmenger syndrome refers to a condition in which progressive pulmonary hypertension leads to a shunt reversal. The subsequent right-to-left shunting of blood creates a condition whereby an increased percentage of blood is ejected from the left ventricle into the systemic circulation without being oxygenated. This phenomenon, assessed via measurement of shunt fraction, refers to the percentage of blood that leaves the heart without being oxygenated. A normal shunt fraction in a healthy individual is approximately 3 to 5 percent. In the presence of a condition that causes the shunt fraction to rise above approximately 20 percent, serious sequelae may ensue. This syndrome is more likely to occur with a VSD or a PDA because of the high pressure and high flow associated with these defects. Poor prognosis associated with patients with Eisenmenger syndrome is associated with arterial oxygen saturation (SaO2) levels less than 85 percent, history of syncope, early onset of clinical deterioration, complexity of CHD, and ventricular dysfunction. Death secondary to Eisenmenger syndrome generally occurs in patients in their 30s and 40s and can be attributed to heart failure, hemoptysis, complications from surgery, and consequences of exercise and pregnancy.36 Decreased SVR associated with normal pregnancy increases the occurrence of right-to-left shunting of blood, resulting in decreased pulmonary perfusion and hypoxemia, thus causing adverse effects for the mother and fetus. Continued pulmonary hypertension may lead to systemic hypotension and decreased right heart filling pressures that may be inadequate to perfuse the pulmonary arterial system. This insufficiency may subsequently result in sudden severe hypoxemia and death.
Women with Eisenmenger syndrome should be counseled with respect to the significant risks that accompany pregnancy. Despite improvements in clinical management, maternal mortality associated with this syndrome has not declined, and is reported to be between 30 and 50 percent.32,37 Termination of pregnancy is generally strongly considered as an option for women with this cardiac disorder. However, there have been reports that note the possibility of improved pregnancy outcome. One such report by Gleicher and colleagues described 13 pregnancies in 12 women with this syndrome who desired to continue their pregnancies, despite recommendations for therapeutic abortion. 38 Three maternal deaths occurred, and eight infants were born alive. In this group of women, successful outcomes were attributed to prolonged bed rest and the use of heparin and oxygen therapy. Saha and colleagues noted 1 postpartum death in a group of 26 pregnant women with Eisenmenger syndrome.39 Smedstad and colleagues reported on the outcomes of a series of eight patients with Eisenmenger syndrome.40 Seven women survived following vaginal deliveries, with follow-up ranging from 3 months to 4 years. The one death occurred in a woman with undiagnosed disease who underwent operative delivery, following the onset of progressive hypoxia and cardiovascular decompensation with concomitant fetal compromise.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


