Calcium, Phosphorus, and Magnesium Metabolism
Allen W. Root
NORMAL MINERAL HOMEOSTASIS
 CALCIUM
CALCIUM
Calcium (Ca) is present largely (99%) in the skeleton as the hydroxyapatite crystal of calcium phosphate [Ca10(PO4)10(OH)2]. Only the 1% to 2% that resides in recently deposited and rapidly exchangeable surface bone, blood, and extracellular fluid is readily mobilized. Total serum calcium is composed of about 50% ionized calcium (Ca2+), the physiologically active form, about 40% calcium bound to albumin and globulins where it is inert, and 10% is complexed to citrate, lactate, bicarbonate, phosphate, or sulfate. Systemic acidosis decreases calcium binding to albumin thus increasing serum Ca2+ levels, while alkalosis increases calcium binding to albumin and lowers Ca2+ values.
Serum Ca2+ levels are maintained within narrow limits by a complex integration of the plasma membrane Ca2+ sensing receptor (CaSR); parathyroid hormone (PTH) and its receptor (PTH/PTH-related protein [PTHrP]-R-PTHR1; calcitonin, the product of the thyroidal parafollicular (C) cell and its receptor; and the vitamin D hormone system acting upon the intestinal tract, bone, and kidney (Fig. 542-1).1-3 As the serum Ca2+ concentration increases, the CaSR on the chief cell of the parathyroid gland (PTG) is activated and decreases PTH synthesis and release. Activation of the CaSR in the distal renal tubule decreases reabsorption of calcium and increases its urinary excretion. PTH stimulates osteoclastic bone re-absorption and increases renal tubular synthesis of 1,25 dihydroxyvitamin D3 (calcitriol) and intestinal absorption of calcium.
Ca2+ enters cells through transmembrane calcium channels. Estrogens and growth hormone (GH) increase, and thyroid hormones, glucocorticoids, vitamin D deficiency, chronic renal disease, and hypoparathyroidism impair intestinal calcium absorption. Currently recommended intakes of calcium range from 200 mg/day in infancy to 1300 mg/day in adolescents (see Table 23-3). 
Calcium is primarily excreted by the kidney and the intestinal tract. After crossing the renal glomerular membrane, 70% of ultrafiltrable serum calcium is reabsorbed in the proximal renal tubule, 20% in the thick ascending loop of Henle, and 8% in the distal convoluted tubule.7 Renal calcium excretion is increased by excessive intake, hypercalcemia of various causes, expansion of extracellular volume, metabolic acidosis, and loop diuretics such as furosemide, glucocorticoids, and mineralocorticoids.
Renal calcium excretion is increased by excessive intake, hypercalcemia of various causes, expansion of extracellular volume, metabolic acidosis, and loop diuretics such as furosemide, glucocorticoids, and mineralocorticoids.
In utero, calcium is actively transported across the placenta under the control of calcitriol and PTHrP. Fetal serum calcium concentrations are high (12 to 13 mg/dL) but quickly fall during the first 24 to 48 hours after delivery in the full-term neonate to a nadir value of 9 mg/dL and then increase to approximately 10 mg/dL. In preterm infants, low-birth-weight (LBW) infants, or full-term ill infants, the fall in calcium concentrations may be exaggerated resulting in hypocalcemia. In the child and adolescent, serum calcium concentrations vary slightly with age and the analytical laboratory (total calcium concentrations: 1 to 5 years, 9.4 to 10.8; 6 to 12 years, 9.4 to 10.2; > 20 years, 8.8 to 10.2 mg/dL.8
The calcium sensing receptor (CaSR) regulates the serum concentration of Ca2+ by determining the rates of secretion and synthesis of PTH and of calcium reabsorption in the renal tubule. In thyroid C cells, increases in serum Ca2+ concentrations release calcitonin, a hypocalcemic peptide. Calcimimetics (eg, cinacalcet) are synthetic compounds that are agonists, and calcilytics are antagonists of the CaSR.10
 PHOSPHORUS
PHOSPHORUS
In vivo, 55% of phosphorus is present as free orthophosphate anions (HPO42– or H2PO4–); 35% is complexed to calcium, sodium, or magnesium; and 10% is bound to protein. Bone hydroxyapatite accounts for 85% of body phosphate stores. The concentration of serum phosphate is governed by its intake, intestinal absorption, excretion, and renal tubular reabsorption. Excessive amounts of calcium and aluminum precipitate intraluminal phosphate and impede its absorption as do primary malabsorption disorders. Phosphate is excreted by the kidney. Renal tubular reabsorption of phosphate is augmented by hypophosphatemia, hypercalcemia, decreased extracellular fluid volume, and metabolic alkalosis. Serum phosphate levels peak in infancy and early childhood and decline during later childhood and adolescence to adult values.
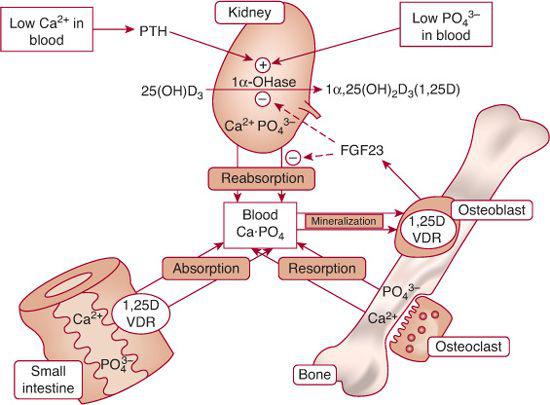
FIGURE 542-1. Regulation of calcium and phosphate homeostasis. In response to calcitriol [1,25(OH)2D3] and parathyroid hormone (PTH), calcium is absorbed from the intestinal tract, kidney tubule, and bone. Calcitonin inhibits resorption of bone calcium. The Ca2+-sensing receptor (CaSR) modulates the activity of the parathyroid glands and the renal tubules. PTH, hypocalcemia, and hypophosphatemia stimulate renal tubular synthesis of calcitriol. PTH inhibits renal tubular reabsorption of phosphate, as does fibroblast growth factor-23 (FGF23), a phosphatonin secreted by osteoblasts. FGF23 also inhibits renal tubular synthesis of calcitriol. VDR, Vitamin D nuclear receptor.
Phosphatonins are phosphaturic substances that inhibit renal tubular phosphate uptake by downregulating expression of SLC34A1.11 They also suppress renal tubular synthesis of calcitriol by inhibiting expression of CYP27B1, thus decreasing intestinal absorption of calcium and phosphate. In osteoblasts and osteocytes, expression and secretion of FGF23 are stimulated by calcitriol.12,13 Serum values of FGF23 are increased in patients with X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, tumor-induced osteomalacia, and fibrous dysplasia. Other phosphatonins are FGF7, matrix extracellular phosphoglycoprotein, and serum frizzled related protein-4.15
 MAGNESIUM
MAGNESIUM
Magnesium (Mg2+) is the second most common cation in the intracellular fluid. It is important for normal bone formation and is an essential component of critical enzymatic reactions including many kinases, ATPase and GTPase. Body magnesium is distributed such that 54% is in the skeleton as a component of bone, 45% is in the intracellular fluid, and only 1% is in the extracellular space. Approximately 0.3% Mg2+ is distributed in plasma; about 30% is bound to protein. Normal serum concentrations of Mg2+ range from 1.5 to 2.3 mg/dL (0.7 to 1.0 mmol/L). Serum concentrations of magnesium in neonates have been reported to be somewhat lower, with total serum magnesium levels of 1.4 to 2.0 mg/dL (0.58 to 0.83 mmol/L) and ionized Mg2+ levels of 0.97 to 1.26 mg/dL (0.40 to 0.56 mmol/L).
Magnesium is ubiquitous in foods, but it is particularly abundant in dairy products, bread, cereals, leafy vegetables, and meat. Intake varies between 200 and 600 mg per day in normal adults. Thirty percent to 50% is absorbed, predominantly in the distal small bowel. Vitamin D enhances intestinal Mg2+ absorption but to a much lesser extent than for Ca2+ absorption. The efficiency of Mg2+ absorption decreases with increasing intake. Absorption increases following magnesium depletion, during periods of rapid growth, and with ingestion of large quantities of phosphate.
Magnesium is primarily excreted by the kidney.  Changes in distal tubular absorption of Mg2+ provide for selective magnesium conservation when intake decreases or if there are increased losses due to intestinal malabsorption. Increased serum Mg2+ or Ca2+ inhibits Mg2+ uptake through activation of the extracellular Ca2+/Mg2+-sensing receptor (CASR).
Changes in distal tubular absorption of Mg2+ provide for selective magnesium conservation when intake decreases or if there are increased losses due to intestinal malabsorption. Increased serum Mg2+ or Ca2+ inhibits Mg2+ uptake through activation of the extracellular Ca2+/Mg2+-sensing receptor (CASR).
 ALKALINE PHOSPHATASE
ALKALINE PHOSPHATASE
Tissue-nonspecific alkaline phosphatase (TNSALP) is synthesized by osteoblasts, liver, kidney, and skin fibroblasts. The osteoblast also synthesizes a specific bone isoform of alkaline phosphatase (BSAP) that is anchored to the osteoblast’s cell surface. BSAP binds to collagen type I and prepares the skeletal matrix for mineralization, increases local concentrations of phosphate by dephosphorylating protein-bound phosphates, enables the transport of inorganic phosphate and Ca2+ into the cell, and inactivates inhibitors of mineralization by removing phosphate groups.
CIRCULATING HORMONES AND RECEPTORS
 PARATHYROID HORMONE
PARATHYROID HORMONE
PTH is synthesized, stored, and secreted by the chief cells of 4 paired parathyroid glands, derived from the third (inferior) and fourth (superior) pharyngeal pouches. The synthesis and secretion of PTH is increased by low and suppressed by high Ca2+ concentrations. Elevated phosphate values increase and low phosphate levels depress PTH production and secretion. High calcitriol concentrations also inhibit PTH synthesis and secretion. Both hypomagnesemia and hypermagnesemia suppress PTH release but not its synthesis. PTH is synthesized as a 115 amino acid propeptide and processed to PTH1-84 that is secreted. In the circulation, the half-life of PTH1-84 is approximately 2 minutes. PTH raises serum calcium concentrations by increasing its renal tubular reabsorption, mobilization from bone, and intestinal absorption, and depresses phosphate levels by inhibiting its renal tubular reabsorption. PTH increases renal tubular synthesis of calcitriol, thus augmenting intestinal absorption of calcium. Acting through the osteoblast, PTH also stimulates osteoclastogenesis and bone resorption.
 PARATHYROID HORMONE-RELATED PROTEIN
PARATHYROID HORMONE-RELATED PROTEIN
PTHrP is a 141-amino-acid peptide that is homologous with PTH at its amino terminal region and is recognized by the type 1 PTH receptor. PTHrP was originally identified as a product secreted by tumor cells that caused “hypercalcemia of malignancy,” a syndrome that can mimic primary hyperparathyroidism PTHrP. It is synthesized in fetal and adult tissues (placenta, PTGs, cartilage, bone, muscle, skin) and functions mainly as a tissue growth and differentiation factor at the local level, and a regulator of smooth muscle tone. It is essential for chondrocyte differentiation and maturation, endochondral bone formation, mammary gland development, tooth eruption, and epidermal and hair follicle growth acting primarily as a paracrine or juxtacrine messenger.16 In the fetus, PTHrP increases calcium transport across the placenta from mother to fetus. PTHrP has effects similar to those of PTH on calcium, phosphate, and vitamin D metabolism. Postnatally, serum levels of PTHrP are low. PTHrP values are high in breast milk.
 PTH/PTHrP RECEPTORS
PTH/PTHrP RECEPTORS
PTH and PTHrP utilize PTHR1 to exert their cellular effects in osteoblasts, renal tubules, skin, and breast. In turn, PTHR1 activates stimulatory G proteins (Gs, Gq) and their respective adenylyl cyclase and phospholipase C signal transduction pathways. After activation of the G-protein, PTHR1 enters the cell through the process of endocytosis where it may be recycled to the cell membrane, guided to the nucleus where it may influence gene transcription, or degraded.
 CALCITONIN
CALCITONIN
Calcitonin is a 32 amino acid hypocalcemic peptide secreted by the C cells of the thyroid gland which lowers serum calcium levels by inhibiting osteoclastic bone resorption.17 The secretion of calcitonin is primarily stimulated by increasing serum concentrations of Ca2+. Calcitonin concentrations are high in the (hypercalcemic) fetus, and fall rapidly after birth, paralleling the decline in serum Ca2+ values. Calcitonin values are relatively constant (< 12 pg/mL) after 3 years of age. However, immunoassays for calcitonin yield inconsistent measurements.
 VITAMIN D
VITAMIN D
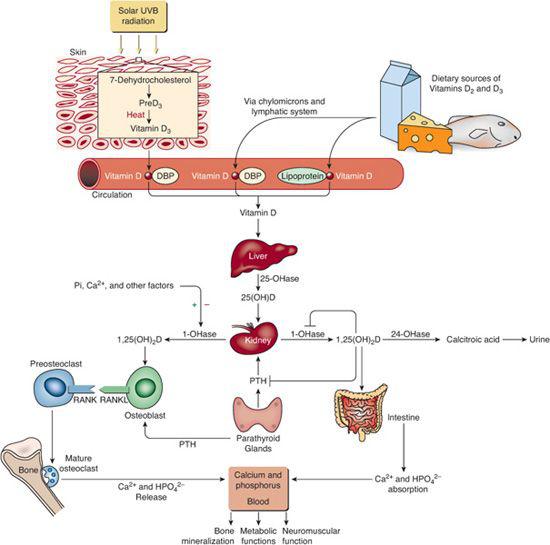
Cholecalciferol (vitamin D3) is a prohormone produced in skin by exposure to sunlight and is also ingested in egg yolk, salmon, herring, and mackerel; ergocalciferol (vitamin D2 ) is derived from plants and yeast18 (Fig. 542-2). The cutaneous synthesis of cholecalciferol is influenced by the latitude, season of the year, and skin pigmentation. After endogenous synthesis or oral ingestion, vitamin D is linked to vitamin D-binding protein and carried to the liver where it is hydroxylated to form 25-hydroxyvitamin D (calcidiol is 25OHD3) by vitamin D-25 hydroxylase (CYP2R1). Bound to vitamin D-binding protein, 25OHD moves to the proximal convoluted and straight renal tubules where it is hydroxylated to the biologically potent metabolite 1,25-dihydroxyvitamin D3 [1,25(OH)2D3 = calcitriol]. Synthesis of 25OHD-1α-hydroxylase is suppressed by increased levels of Ca2+ and phosphate, FGF23, and indirectly by calcitriol through inhibition of PTH secretion. Bone, intestine, liver, and kidney inactivate calcitriol and increase its excretion in urine and bile. Many drugs (eg, phenobarbital, phenytoin, carbamazepine) increase hydroxylation of calcitriol and its urinary excretion.
) is derived from plants and yeast18 (Fig. 542-2). The cutaneous synthesis of cholecalciferol is influenced by the latitude, season of the year, and skin pigmentation. After endogenous synthesis or oral ingestion, vitamin D is linked to vitamin D-binding protein and carried to the liver where it is hydroxylated to form 25-hydroxyvitamin D (calcidiol is 25OHD3) by vitamin D-25 hydroxylase (CYP2R1). Bound to vitamin D-binding protein, 25OHD moves to the proximal convoluted and straight renal tubules where it is hydroxylated to the biologically potent metabolite 1,25-dihydroxyvitamin D3 [1,25(OH)2D3 = calcitriol]. Synthesis of 25OHD-1α-hydroxylase is suppressed by increased levels of Ca2+ and phosphate, FGF23, and indirectly by calcitriol through inhibition of PTH secretion. Bone, intestine, liver, and kidney inactivate calcitriol and increase its excretion in urine and bile. Many drugs (eg, phenobarbital, phenytoin, carbamazepine) increase hydroxylation of calcitriol and its urinary excretion.
Acting through the nuclear vitamin D receptor (VDR), the major effect of calcitriol is to increase intestinal and renal absorption of calcium and phosphate and to maintain normal serum concentrations of these ions by enhancing expression of genes that encode calcium transporters and channels. Calcitriol also stimulates formation of cortical, trabecular, and endochondral bone.
Serum concentrations of 25OHD reflect body stores of vitamin D. Currently defined normal serum 25OHD levels (10 to 55 ng/mL) are likely too low; a more physiologic range of normal values is 32 to 80 ng/mL. Normal ranges of calcitriol concentrations are as follows: neonates 8 to 72 pg/mL; infants and children 15 to 90 pg/mL; and adults 21 to 65 pg/mL. Current recommendations for daily oral vitamin D intake range from 200 IU in infants to 400 IU in children and adolescents to 1000 IU during pregnancy and lactation, but there is substantial evidence to indicate that these doses are inadequate and that there is a high incidence of subclinical vitamin D deficiency.19,20
 VITAMIN D RECEPTOR (VDR)
VITAMIN D RECEPTOR (VDR)
The VDR is found in the intestinal tract, distal renal tubule, osteoblast, keratinocyte, hair follicle, fibroblast, muscle, thyroid, PTG, pancreas, placenta, activated T and B lymphocytes, macrophages, and monocytes. The synthesis of the VDR is stimulated by calcitonin, retinoic acid, estrogen, and β-catenin. After binding to calcitriol, the VDR pairs with the retinoid X receptor (RXRα) to form a heterodimer that binds the target genes  .
.
HYPOCALCEMIA
Hypocalcemia is the consequence of either too little calcium entering the circulation from the gastrointestinal tract, bone, or kidney, or its excessive loss into urine, stool, or bone. The causes of hypocalcemia are most easily considered when divided into neonatal causes, hypoparathyroidism, vitamin D deficiency, and miscellaneous disorders as listed in Table 542-1. These are discussed below. The approach to evaluation and management is then discussed. A large number of disorders due to specific gene mutations are also associated with disorders of mineral and skeletal homeostasis and are shown in eTable 542.1  .
.
 DISORDERS ASSOCIATED WITH NEONATAL HYPOCALCEMIA
DISORDERS ASSOCIATED WITH NEONATAL HYPOCALCEMIA
Causes of neonatal hypocalcemia include a blunted post natal secretion of PTH, decreased renal tubular excretion of phosphate in response to PTH with resultant hyperphosphatemia, excessive and prolonged secretion of calcitonin, hypomagnesemia, resistance to the intestinal calcium absorptive and skeletal calcium reabsorptive actions of calcitriol, and rapid deposition of calcium into bone.
Hypocalcemia developing within the first 72 hours after delivery (early hypocalcemia) occurs in full-term ill newborns, low-birth-weight (both premature or small-for-gestational-age) neonates, and in those born to women with vitamin D deficiency, diabetes mellitus, chronic renal disease, or other illnesses. If the mother had hyperparathyroidism or ingested large quantities of calcium-containing antacids during gestation, increased placental transfer of calcium suppresses PTH secretion, and return of normal parathyroid function after birth may require weeks or months.22-52
Hypocalcemia appearing after 72 hours of age (late neonatal hypocalcemia) results from increased dietary intake of calcium-binding agents such as phosphate (evaporated milk or modified cow milk formulas) or fiber, hypomagnesemia, hypoparathyroidism, or vitamin D deficiency. Hyperphosphatemia and hypocalcemia may be due to hypoparathyroidism if PTH levels are low, or to renal insufficiency (renal hypoplasia or obstructive uropathy) if PTH and creatinine values are elevated. Neonatal vitamin D deficiency is characterized by hypocalcemia, hypophosphatemia, and elevated alkaline phosphatase. It occurs as a consequence of maternal vitamin D deprivation (especially in the older breastfed infant with a vegetarian mother and who has little sun exposure), altered activity of renal 25-hydroxyvitamin D-1 hydroxylase, or inactivating mutations of the vitamin D receptor.53 At 3 to 4 months of postnatal age, if the LBW infant has only marginal ingestion of calcium, phosphate, and vitamin D, the infant can develop hypocalcemia with osteopenia due to calcium depletion.
 HYPOPARATHYROIDISM
HYPOPARATHYROIDISM
Hypoparathyroidism may be due to dysgenesis of the parathyroid glands or to their destruction by inflammatory, infiltrative, surgical, or radiation insults, to errors in the synthesis of PTH, or to abnormalities of tissue responsiveness to PTH.54,55 In neonates and infants, hypoparathyroidism is often transient due to delayed maturation of parathyroid gland activity and resolves a few weeks after birth.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


