Brain Abnormalities and Injuries
Seetha Shankaran and C. Michael Cotton
Neonatal encephalopathy is a clinically defined syndrome of disturbed neurological function in the early postnatal days of life in the term infant, manifested by a combination of signs including altered consciousness, abnormal muscle tone or reflexes, altered respiration or seizures. Etiologies of neonatal encephalopathy include the combination of intrapartum or antepartum hypoxia and ischemia (hypoxic-ischemic encephalopathy), which may be accompanied by some prenatal signs of fetal distress; vascular pathologies, including intracranial bleeding and stroke; injuries secondary to birth trauma; infections; genetic and metabolic disorders; and congenital brain abnormalities. This chapter is focused on brain injuries and abnormalities in term newborn infants, with particular emphasis on infants who present with biochemical and clinical evidence of hypoxic-ischemic encephalopathy, and the current diagnostic and treatment approaches to such injury. Other birth injuries associated with central nervous system damage including vascular malformations and birth trauma are briefly discussed as well.
EPIDEMIOLOGY OF ENCEPHALOPATHY
Neonatal encephalopathy occurs in 1 to 6 per 1000 live full-term births, with a recent population-based estimate of 1.9 to 3.8 per 1000.1 Fifteen to 20% of affected newborns will die in the postnatal period, and an additional 25% will sustain childhood disabilities.2 Neonates with mild encephalopathy do not have an increased risk of motor or cognitive deficits. Neonates with severe encephalopathy have an increased risk (> 60%) of death or of cerebral palsy and mental retardation. Neonates with moderate encephalopathy have a higher likelihood of death or deficits, such as memory impairment, visual motor or visual perceptive dysfunction, increased hyperactivity, and delayed school readiness, that is approximately half that of those with severe encephalopathy.
The cause and timing of such injuries is usually unknown. Since neonatal encephalopathy has myriad causes, diagnostic criteria that suggests a hypoxic-ischemic insult is attributable to an acute intrapartum event has been suggested. These include metabolic acidosis with a cord pH below 7 or a base deficit of 12 mmol/L or greater, early onset of encephalopathy, and exclusion of another etiology such as trauma, coagulation disorder, and genetic and metabolic causes. Infants with severe injury and hypoxic-ischemic encephalopathy related to an intrapartum event develop either spastic quadriplegia or dyskinetic type cerebral palsy. Signs consistent with an event in the 48 hours prior to delivery, but not necessarily an acute event, include a sentinel event occurring immediately before or during labor; a sudden sustained fetal bradycardia or absence of fetal heart rate variability in the presence of persistent, late, or variable decelerations, usually after a sentinel event before which the fetal heart rate pattern was normal; Apgar scores less than 3 at 5 minutes; multisystem organ involvement apparent within 72 hours of birth; and early imaging studies demonstrating evidence of acute nonfocal cerebral abnormalities.3
The vast majority of infants with encephalopathy do not have an identifiable intrapartum event such as cord prolapse or uterine rupture.4 Although there is no specific diagnostic test for hypoxic-ischemic encephalopathy, neuroimaging studies identifying injury to the basal ganglia and parasagittal white matter suggest an injury that occurred to tissues susceptible to hypoxic-ischemic injury in the perinatal period rather than long-standing antenatal compromise or an acute vascular event. Although the neuroimaging results may identify perinatal injury, precise timing of perinatal injury by neuroimaging alone is not possible.
PATHOPHYSIOLOGY OF HYPOXIC-ISCHEMIC BRAIN INJURY
The pathophysiology of brain injury secondary to hypoxia-ischemia is simplified into 2 phases of pathologic events that culminate in sustained brain injury. These phases are primary and secondary energy failure based on characteristics of the cerebral energy state used to describe the temporal sequence in newborn animals.5,6 Primary energy failure is characterized by reductions in cerebral blood flow and delivery of oxygen and substrates to brain tissue. High-energy phosphorylated compounds such as adenosine triphosphate and phosphocreatine are reduced, and tissue acidosis is prominent. This phase is an essential prerequisite for all deleterious events that follow. Primary energy failure is associated with acute intracellular derangements, such as loss of membrane ionic homeostasis, excessive release or blocked reuptake of excitatory neurotransmitters, defective osmoregulation, and inhibition of protein synthesis. Excessive stimulation of neurotransmitter receptors and loss of ionic homeostasis mediate an increase in intracellular calcium and osmotic dysregulation. Elevation in intracellular calcium triggers a number of destructive pathways by activating lipases, proteases, and endonucleases.
Resolution of hypoxia-ischemia within a specific time interval reverses the fall in high-energy phosphorylated metabolites and intracellular pH and promotes recycling of neurotransmitters. The therapeutic window for hypoxia-ischemia to be successfully reversed and to promote recovery is influenced by maturation, preconditioning events, substrate availability, body temperature, and coexisting disease processes. Although recovery of the cerebral energy state may occur following primary energy failure, a second interval of energy failure may occur at a time remote from the initiating event. Secondary energy failure differs from primary energy failure in that declines in phosphocreatine and adenosine triphosphate are not accompanied by brain acidosis. The presence and severity of secondary energy failure depends on the extent of primary energy failure. The pathogenesis of secondary energy failure is not as well understood as that of primary energy failure but likely involves multiple pathophysiologic processes, including accumulation of excitatory neurotransmitters, oxidative injury, apoptosis, inflammation, and altered growth factors and protein synthesis. Much of the later injury occurs secondary to the process of apoptosis, or programmed cell death. Apoptosis occurs in normal brain development and is useful for refining cell connections and pathways. The cellular signals after hypoxic-ischemic injury accelerate this process in normal brain tissue, contributing significantly to the later evolving injury noted in infants with hypoxic-ischemic encephalopathy.
The interval between primary and secondary energy failure represents a latent phase that corresponds to a potential therapeutic window, as clinicians usually do not know the precise time of primary energy failure. Initiation of therapies during this latent phase in perinatal animals has been successful in reducing brain damage, substantiating the concept a therapeutic window. The duration of the therapeutic window is approximately 6 hours in near-term fetal sheep based on the neuroprotection associated with brain cooling initiated at varying intervals following brain ischemia.
DIAGNOSIS OF ENCEPHALOPATHY
Neonatal encephalopathy is characterized by the presence of an abnormal neurologic examination in the first postnatal days, ranging from lethargy, hypotonia, or hypertonia to a normal appearance with the sudden occurrence of apnea or seizures, which can be subtle and subclinical, focal, or generalized and clinically obvious.7 The clinician also must be aware that many infants present with myoclonus, which may appear to be seizures. In extreme cases, an otherwise apparently well-grown, healthy term neonate may present with coma after extremely low Apgar scores and severe metabolic acidosis on cord blood gases.
The first step in diagnosis of neonatal encephalopathy is to obtain a detailed history of the pregnancy and intrapartum period. Any event likely to compromise blood or oxygen supply to the fetus, such as placental abruption, uterine rupture, amniotic fluid embolism, tight nuchal cord, cord prolapse or avulsion, maternal hemorrhage, trauma or cardiorespiratory arrest, severe and sustained fetal bradycardia, or prolonged labor, may be causative. Most infants with encephalopathy do not have an obvious cause for the encephalopathy. A history of maternal elevation of temperature has prognostic significance that increases the risk of neonatal encephalopathy and cerebral palsy. A history of fetal tachycardia and maternal tachycardia may also raise suspicions of chorioamnionitis. The placenta should be examined to determine if there was placental infection or noninfectious etiologies for hypoxic-ischemic encephalopathy.
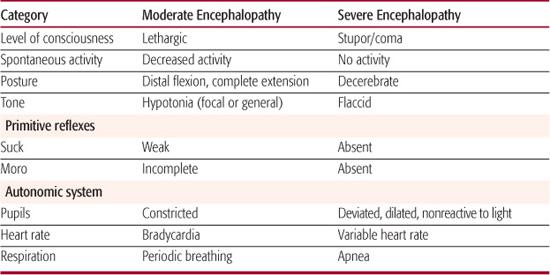
All neonates with encephalopathy should have detailed neurologic examinations repeatedly during the first postnatal days to evaluate the presence of mild, moderate, or severe encephalopathy based on the Sarnat classification7 (Table 52-1). The physical examination, acid-base data, intrapartum history and Apgar scores provide useful indicators of status. Ongoing encephalopathy through the first postnatal week, along with serial neuroimaging and continuing burst suppression on electroencephalograph, can be considered poor prognostic signs. Neuroimaging with magnetic resonance imaging, particularly with added diffusion weighted imaging, which measures diffusion of water in tissues (less apparent diffusion noted using this methodology is proportional to more injury), in the first week of life and then repeated during the postnatal months is also helpful in gaining understanding about the extent and etiology of injury. In adults, biomarkers such as various inflammatory cytokines are beginning to be used to identify individuals with stroke and to guide therapy. The use of these biomarkers to grade injury, guide therapy, and predict outcome in neonatal encephalopathy is currently being investigated.
CEREBRAL MONITORING
The best current bedside tool for cerebral function monitoring in term and near-term infants is the amplitude integrated EEG (aEEG), which correlates well with conventional EEG. The aEEG records a single-channel EEG from biparietal electrodes; the signal is then filtered, rectified, and smoothed, and amplitude is integrated. The aEEG interpretation is based primarily on pattern recognition. It appears to be predictive of neurodevelopmental outcome in term infants with hypoxic-ischemic encephalopathy, and coupled with an early neurologic examination, the aEEG correlates well with persistent encephalopathy. It has been suggested that aEEG should become part of the initial evaluation of near-term and term infants with hypoxic-ischemic encephalopathy. Additionally, early work combining analysis of regional cerebral oxygen saturation and fractional cerebral tissue oxygen extraction (FTOE) measured by near-infrared spectroscopy in infants with hypoxic-ischemic encephalopathy suggests the aEEG plus the FTOE are somewhat predictive of outcome, and the regional saturation and the FTOE can identify the period of secondary injury and energy failure that may be our best target for therapeutic intervention. The aEEG should not be used for the detection and treatment of neonatal seizures because it has not been proven to reliably detect subclinical seizures. Cerebral oximetry is also early in its development for use in therapeutic decision making in the intensive care nursery.
CURRENT THERAPIES FOR HYPOXIC-ISCHEMIC ENCEPHALOPATHY
The management of neonates with hypoxic-ischemic encephalopathy has been limited to generally supportive intensive care that includes correction of hemodynamic and pulmonary disturbances (hypotension and hypoventilation), correction of metabolic disturbances (related to glucose, calcium, magnesium, and electrolytes), treatment of seizures, and monitoring for other organ system dysfunction, especially hepatic, renal, and coagulation status. Avoidance of hyperthermia may also be beneficial, and monitoring of urine output can guide fluid maintenance and drug dosing. This management approach is directed at avoiding injury from secondary events associated with hypoxia-ischemia. Over the past 15 years specific therapies to block or dampen the cascade of events triggered by hypoxia and ischemia, and to prevent or treat neonatal seizure have been evaluated. These include the use of anticonvulsants, and brain hypothermia.
Treatment of Seizures Because neonatal seizure may have adverse effects, their prevention and treatment with prophylactic administration of anticonvulsants has been considered. However, the consequences of subclinical or electroencephalogram detected neonatal seizures on neurodevelopmental outcome has not been adequately evaluated. There is no current data that demonstrates treatment with anticonvulsants or barbiturates can effectively control seizures or that treatment with these agents reduces cerebral palsy or mental retardation in infants with encephalopathy. The existing studies are small and there is concern that treatment may have adverse effects on the central nervous system. Therefore, larger scale clinical trials of anticonvulsant and barbiturate therapy are needed before these agents becomes widespread.
Brain Hypothermia In experimental models of brain ischemia in neonatal animals, it has been demonstrated that a small reduction in brain temperature (1–6°C) during or immediately after ischemia decreases the release of excitatory neurotransmitters, caspase 3 activation, decreases apoptosis, and blunts the usually observed decrease in protein synthesis, increase in free radicals and changes in microglia activation and cytokine production.
These findings justified clinical trials. To date, there have been 2 large, randomized, controlled trials and 1 large pilot study to evaluate the efficacy of hypothermia as neuroprotection for term and near-term infants with hypoxic-ischemic encephalopathy.
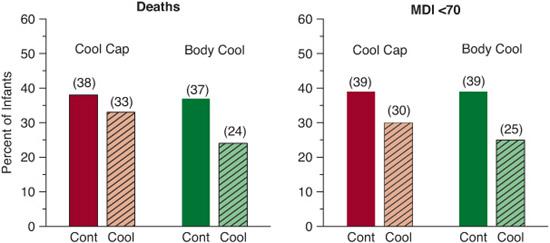
The multicenter CoolCap study involved 243 infants, with moderate or severe encephalopathy and an abnormal aEEG, who were either cooled to a core body temperature of 34 to 35°C for 72 hours or treated with temperature maintenance in normothermia range with conventional care. The primary outcome of the study was death or disability at 18 months. Cooling was provided by selective head cooling with mild systemic cooling. Death or severe disability occurred in 66% of infants randomized to conventional care and 55% randomized to the cooled group: odds ratio (95% CI) 0.61 (0.34 – 1.09), P = 0.10.8 The effect of head cooling for infants with the most severe aEEG changes was not protective; on the other hand, the effect of head cooling for infants with less severe aEEG changes (n = 172) was protective: odds ratio 0.42 (0.22 – 0.80; P = 0.009).
Table 52-1. Stages of Encephalopathy per Sarnat Classification
One smaller randomized, controlled pilot study performed at 7 centers with 65 infants involved moderate systemic whole-body hypothermia to 33°C for 48 hours compared to normothermia maintained at 37°C.9 The safety report of this pilot study documented that infants in the hypothermia group had more significant bradycardia, longer dependence on pressor medications, higher prothrombin times, more seizures, and need for more plasma and platelets transfusions. At 12 months of age, death or severe motor disability occurred in 52% of hypothermia group compared to 84% of normothermia group (P = 0.02), but the normothermia group lacked follow up information on 8 of 33 enrolled infants versus only 5 of 32 for the treated group. In a subgroup analysis, outborn infants were more likely than inborn infants to die: odds ratio 10.7 (1.3 – 90.0).
The National Institute of Child Health and Human Development (NICHD) Neonatal Research Network trial evaluated 102 infants randomized to hypothermia with whole-body cooling to 33.5°C for 72 hours compared to 106 control infants randomized to conventional care.10 The primary outcome, death or moderate to severe disability at 18 months of age, was noted in 44% of infants in the hypothermia group compared to 62% of infants in the control group with a risk ratio of 0.72 (0.54 – 0.95). There was a trend for cooling to benefit infants in both moderate and severe encephalopathy groups.
Other secondary analyses have been published from the NICHD randomized clinical trial. In one study, examining the relationship of elevated temperature after hypoxic-ischemic encephalopathy, 22% of esophageal core temperatures measured among the control group infants were higher than 37.5°C.11 The odds of death or disability were increased 3.6-fold to 4-fold for each centigrade increase in the highest quartile of temperature in the control group. These results may reflect underlying brain injury and/or adverse effects of high core temperature on outcome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


