Blood
Lori Luchtman-Jones
Washington University School of Medicine, St. Louis Children’s Hospital, St. Louis, Missouri 63110.
NORMAL HEMATOPOIESIS
Hematopoiesis in the human embryo and fetus can be divided into three overlapping phases: mesoblastic, hepatic, and myeloid (1). The earliest cells of erythroid lineage have been detected at day 14 of gestation within the blood islands of the yolk sac. By day 35, erythropoiesis occurs in the liver, and granulopoiesis and thrombocytopoiesis follow. Until 6 months gestation, the liver is the major site of hematopoiesis. The myeloid phase starts in the fourth month, when hematopoiesis, including monocyte and lymphocyte production, begins in the bone marrow. The bone marrow becomes the major site of hematopoiesis. Through early childhood, most of the bone marrow space is occupied by red (hematopoietically active) marrow. With age, the distal marrow spaces slowly fill with fat.
Fetal erythrocytes differ from those produced in infancy and adulthood (Fig. 16-1). Differences in mean corpuscular volume, membrane properties, hemoglobin content, metabolic profile, and life span have all been well described. In the newborn infant, 70% to 90% of the hemoglobin is of fetal type, which is gradually replaced by adult hemoglobin during the next few months. Hemoglobinopathies characterized by abnormalities of the α chain or embryonic or fetal hemoglobins result in abnormalities of the red blood cells at birth, but abnormalities of the β chain of hemoglobin are detectable later in the first year of life. Because of the lower oxygen saturation of fetal blood, coupled with the relatively poor oxygen release to the tissues by fetal hemoglobin, the hematocrit is elevated in the immediate newborn period relative to later in infancy. The normal ranges for hematocrit, mean corpuscular volume, and reticulocyte count vary depending on the age of the patient, and it is important to consult a standardized chart of age-appropriate values (Table 16-1). The leukocyte count and differential change markedly from birth to infancy to childhood and on to adulthood. Platelet counts remain stable from the birth of a full-term infant to adulthood.
ANEMIA
The usual definition of anemia is a hematocrit value lower than two standard deviations below the mean for age and sex. Because the primary function of the red cell is to transport oxygen from the pulmonary bed to other tissues for release, anemia may impair oxygen delivery. Tissue oxygenation depends on the hematocrit, the oxygen affinity of hemoglobin, and the cardiorespiratory status of the patient. The only absolute indications for rapid correction of anemia by red cell transfusion are to restore tissue oxygenation and to expand blood volume after acute loss. Unfortunately, little information exists about the level of hematocrit necessary for adequate delivery of oxygen to the tissues. Observations on changes in coronary sinus oxygen saturation in dogs with varying degrees of acutely induced anemia (2) and the surgical experience among Jamaican patients with sickle cell disease (3) suggest that otherwise healthy young adults can tolerate short-term anemia with a hematocrit value between 20% and 25% without adverse outcome. In many pediatric centers, the sicker patients, particularly those with cardiorespiratory dysfunction, undergo transfusion to maintain the hematocrit closer to normal and maximize tissue oxygenation.
Causes of anemia include decreased red cell production, accelerated loss or destruction of red cells, or both. Red cell abnormalities associated with early destruction can be congenital or acquired, and intrinsic (e.g., abnormal hemoglobin) or extrinsic (e.g., immune-mediated hemolysis). Anemias can also be differentiated by the indices and morphologic appearance of red cells. Using the latter method, anemia is classified as microcytic, normocytic, or macrocytic, based on the mean corpuscular volume (Table 16-2). Hypochromicity, abnormal red cell shapes, polychromasia, and cellular inclusions such as stippling or Howell-Jolly bodies can provide valuable etiologic clues.
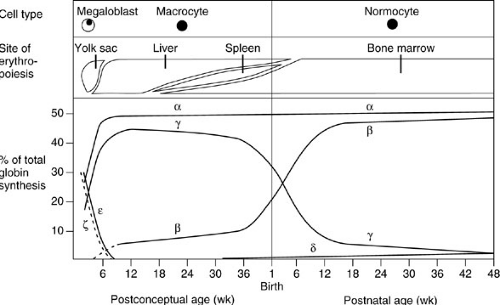 FIGURE 16-1. Alterations in the production site, hemoglobin composition, and morphology of red blood cells during the prenatal and early postnatal periods. (Adapted from Weatherall DJ, Clegg JB. The thalassemia syndromes, 3rd ed. Cambridge: Blackwell Scientific, 1981:64) |
The investigation of anemia begins with a careful patient history, including diet, details of prior anemia or transfusions, pica, evidence for blood loss, medications, and toxic exposures. Family history of anemia, transfusions, gallbladder disease, or splenectomy is also helpful. The physical examination should identify dysmorphic or skeletal abnormalities and document appropriate growth, development, and general health. Cardiopulmonary abnormalities, hepatosplenomegaly, or jaundice should be documented. Stool specimens may be tested for occult blood. Laboratory tests include a complete blood count with red cell indices, a reticulocyte count, and evaluation of the blood smear. The results of this preliminary workup dictate the need for further laboratory testing.
Iron-Deficiency Anemia
Although the incidence of iron-deficiency anemia in the United States has decreased, it remains the leading cause of anemia in infancy and childhood. Factors associated with the decline in the incidence of iron-deficiency anemia include the increasing number of infants receiving breast milk, the extensive use of iron-supplemented infant formulas and baby cereals during the first year of life, and the avoidance of the introduction of cow milk in the infant’s diet until 9 to 12 months of age (4).
The amount of iron stored in the body of the newborn infant is proportional to body weight and, for the full-term infant, should be sufficient for the first 4 to 6 months of life. Preterm infants, however, weigh less at birth, have a much more rapid rate of postnatal growth, and often develop iron deficiency within the first 2 months of life unless their diets are supplemented with iron. Infants who experience accelerated iron losses secondary to blood loss from repeated laboratory testing, trauma, surgery, or anatomic abnormalities also become iron deficient if supplementation is not instituted early. The American Academy of Pediatrics suggests that full-term infants receive, through diet or supplement, at least 1 mg per kg of iron daily from age 4 months to 3 years. Premature or low birth weight infants should be supplemented with 2 mg per kg per day (up to a maximum of 15 mg per day) from 2 months of age, and very low birth weight (VLBW) infants (those weighing less than 1,000 g at birth) should receive 4 mg per kg per day (up to a maximum of 15 mg per day) from the age of 2 months. The higher rates of iron supplementation should be continued through the first year of life (4,5).
Toddlers who are finicky eaters can derive a substantial amount of their daily calories from cow milk, which is a poor source of iron. Adolescence is another period of rapid growth during which dietary intake of iron-containing foods is poor. Iron-deficiency anemia can occur at any time when rapid growth outstrips the ability of diet and body stores to supply iron requirements. Evaluation of dietary iron intake, sources of iron loss, and growth requirements is part of the diagnostic workup of anemia. After infancy, the possibility of iron-deficiency anemia secondary to blood loss or malabsorption becomes increasingly likely.
Early in iron depletion, the bone marrow iron stores decline and ferritin decreases. Subsequently, transferrin saturation drops and serum transferrin receptor levels rise. As iron deficiency develops, serum iron levels drop. Finally, erythrocyte production is affected. A hypochromic, microcytic anemia with a large red blood cell distribution width becomes apparent, accompanied by elevated total iron-binding capacity. Mild deficiency can be difficult to diagnose in patients with acute or chronic illness. Occasionally, the results of the screening tests are inconclusive, and in these cases, a therapeutic trial of iron can be undertaken. Ferrous sulfate in a dose of 3 to 6 mg per kg per day of elemental iron q.d. or divided t.i.d. can be given for 1 month (4). If the hemoglobin rises by at least
1 g per dL in 1 month (in the absence of ongoing blood loss), the diagnosis of iron deficiency is made, and the iron supplementation should be continued until 2 or 3 months after the hemoglobin and hematocrit are in the normal range. Response to iron therapy, measured by elevation of the reticulocyte count, can be observed within 2 to 3 days of the initiation of treatment. Recurrent anemia is most likely due to unrecognized blood loss.
1 g per dL in 1 month (in the absence of ongoing blood loss), the diagnosis of iron deficiency is made, and the iron supplementation should be continued until 2 or 3 months after the hemoglobin and hematocrit are in the normal range. Response to iron therapy, measured by elevation of the reticulocyte count, can be observed within 2 to 3 days of the initiation of treatment. Recurrent anemia is most likely due to unrecognized blood loss.
TABLE 16-1 Age-Specific Values for Complete Blood Count. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 16-2 Morphologic Approach to Diagnosis of Anemia.a | |||
|---|---|---|---|
|
Hemolytic Anemias
Accelerated destruction of red blood cells is the end point of a host of causes of hemolytic anemia. Clinical signs and symptoms may include jaundice, splenomegaly, cholelithiasis, anemia, reticulocytosis, and hemoglobuinuria. The peripheral blood smear may show characteristic changes in the red blood cell morphology. An inappropriately low reticulocyte count indicates suppression of red cell production, most commonly due to infection. Extrinsic influences on the red blood cell include immune hemolysis, exposure to extreme heat, and destruction of red cells by turbulent flow through cardiac valves (Waring blender syndrome) or fibrin-strewn vasculature (microangiopathic hemolysis). A variety of intrinsic abnormalities can affect the life span of the red blood cell, including disorders of the red cell membrane, enzymatic defects, and certain hemoglobinopathies. Folic acid supplementation is recommended in patients with chronic hemolysis to support increased bone marrow activity.
Alloimmune Hemolytic Anemia
Rh disease of the newborn and ABO hemolytic disease are the most common types of alloimmune disease in neonates. Alloimmune hemolysis can occur when the mother lacks an antigen on her red cells such as D (Rh negative) or A and B (type O). If the fetus has an antigen recognized as foreign when fetal red cells enter the maternal circulation, mother may mount an immune response. Maternal IgG can cross the placenta and enter the fetal circulation, where hemolysis may result. Transfusion of incompatible red cells will result in hemolysis in older patients. A more recently recognized type of alloimmunization occurs when donor lymphocytes transplanted in solid organs produce antibodies against recipient red cells.
Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia (AIHA), characterized by the production of antibodies against one’s own red cells, is relatively rare in pediatric patients. Classically, IgG-coated red cells are destroyed in the spleen, and to a lesser extent, in the liver. AIHA may be self-limited in association with infection, drugs, or toxins, but may also be chronic in association with a collagen vascular disorder. The direct antiglobulin test will be positive for IgG, C3, or both
deposited on the red cell membranes. Approximately 80% of IgG-induced AIHA patients will respond to steroid therapy. Splenectomy is effective in cases in which the spleen is the primary site of red cell destruction. The more rare cold agglutinin IgM antibodies cause red cell destruction, in association with complement, predominantly in the liver or, occasionally, in the vasculature. Steroid therapy is generally less effective than in IgG-mediated disease.
deposited on the red cell membranes. Approximately 80% of IgG-induced AIHA patients will respond to steroid therapy. Splenectomy is effective in cases in which the spleen is the primary site of red cell destruction. The more rare cold agglutinin IgM antibodies cause red cell destruction, in association with complement, predominantly in the liver or, occasionally, in the vasculature. Steroid therapy is generally less effective than in IgG-mediated disease.
Hereditary Spherocytosis
A classic example of a nonimmune hemolytic anemia is hereditary spherocytosis (7). Defects in red cell membrane proteins have been described that reduce the stability and deformability of the cytoskeleton. Autosomal dominant, recessive, and dominant inheritance with reduced penetrance have been described. Most patients are identified in childhood. The presenting features, in order of frequency, are spherocytic anemia, jaundice, splenomegaly, and positive family history. Most patients do not have symptoms except for splenomegaly and mild hemolysis. Complications include aplastic crisis, hemolytic crisis, and gallbladder disease. The management of asymptomatic gallstones may be observation or elective cholecystectomy. Patients with abdominal pain and substantial anemia are candidates for splenectomy to prevent premature destruction of red cells. Splenectomy is usually deferred until after 5 years of age because the risk of postsplenectomy sepsis is especially high in infants and young children.
Glucose-6-phosphate Dehydrogenase Deficiency
The most common red cell enzyme defect associated with hemolysis is glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Affected red cells are less able to repair oxidative damage because of lower levels of nicotinamide adenine dinucleotide phosphate (NADPH) production; a shortened red cell life span results. The disorder is transmitted by X-linked inheritance. Three variants of the disease are associated with different geographic locations and variable severity. The Mediterranean variant is found in the Middle East and India. A milder type, G-6-PD A, is common in Africa, southern Italy, Spain, and Mexico. G-6-PD Mahidol occurs in Southeast Asians. The geographic pattern of distribution of this enzyme deficiency parallels that of the prevalence of malaria, leading to the observation that G-6-PD deficiency is somewhat protective from malaria. Acute hemolysis can be triggered by oxidative stress from drugs, chemicals, infection, or fava beans. Hemolysis occurs within 24 to 48 hours of exposure and is accompanied by abdominal pain, vomiting, diarrhea, low-grade fever, jaundice, splenomegaly, hemoglobinuria, and anemia. Management includes discontinuation of the precipitating agent, hydration, and transfusion, if needed. Common drugs to be avoided or used with caution in this disorder include antimalarials, sulfonamides and sulfones, nitrofurans, antihemintics, ciprofloxacin, methylene blue, acetominophen, aspirin, and vitamin K analogues.
Sickle Cell Anemia
In the United States, the incidence of sickle cell disease is about 8% in the African American population. Sickle cell trait (hemoglobin AS), occurring when one normal β-globin gene coding for hemoglobin A and one mutant gene coding for hemoglobin S are inherited, is not associated with disease. Carrier status has been associated with an increased incidence of hematuria and reports of sudden death related to high altitude and extreme exercise. Sickle cell anemia (hemoglobin SS) occurs with the inheritance of two abnormal β-globin genes. There are a number of other sickle hemoglobinopathies in which one gene coding for hemoglobin S is inherited along with a second abnormal β-globin gene coding for another hemoglobin, such as hemoglobin C or β-thalassemia. The clinical courses of disease in these patients may be indistinguishable from, or milder than, the course of hemoglobin SS disease.
The clinical hallmarks of the disease are hemolytic anemia with reticulocytosis and the appearance of irreversibly sickled cells on the peripheral blood smear. Tremendous clinical variability exists, but most patients suffer from painful vasoocclusive crises. Chronic vasculopathy and tissue ischemia result in dysfunction of many organs, including the heart, kidneys, lungs, skin, and central nervous system (CNS). After early childhood, irreversible organ damage is the most frequent cause of death. Acute chest syndrome, characterized by chest pain associated with a new infiltrate on chest radiograph, can progress rapidly to respiratory failure and death. Because of the onset of splenic dysfunction in early childhood, bacterial infections have been a major cause of mortality and morbidity. Penicillin prophylaxis, immunization against Streptococcus pneumoniae and H. influenzae, newborn screening, and improvements in supportive care have contributed to lower morbidity and mortality rates in young children. Splenic sequestration crises, caused by trapping of red blood cells in the spleen, are characterized by abdominal pain, splenomegaly, reticulocytosis, worsening anemia, thrombocytopenia, and decreased circulating blood volume. Life-threatening or recurrent sequestration is an indication for splenectomy. Infections do not appear to be increased in patients with sickle hemoglobinopathies in the first 5 years postsplenectomy (8). Priapism is a distressing, often recurrent problem that causes sexual dysfunction in almost one-half of affected patients.
Most patients with a sickle hemoglobinopathy will receive a blood transfusion during their lifetime, usually in an attempt to improve oxygen delivery and minimize organ dysfunction. Because of the many intrinsic abnormalities
in cells containing hemoglobin SS, these cells have increased viscosity. Increasing the hematocrit by simple transfusion can worsen oxygen delivery to the tissues by raising blood viscosity. In general, anemia below the patient’s baseline level can be treated by simple transfusion. Red cell exchange transfusion is offered if rapid correction is needed in a patient with borderline cardiac function or volume overload, or if management of stroke or acute chest syndrome is desired (9). Although perioperative morbidity and mortality rates in sickle cell disease are improving, the potential for serious complications remains. Standard practice is to offer red cell transfusion to raise the hemoglobin to 10 g per dL with or without reducing hemoglobin SS levels to 30% for major surgical or ophthalmologic procedures. In addition to the usual risks of blood transfusion, these patients have problems with hyperviscosity and iron overload. Alloimmunization rates in patients with sickle cell disease are higher than those in the general population, approaching 20% in most series (Table 16-3). Historically, the most common antigens are Rhesus (Rh), Kell (K), Duffy (Fy), and Kidd (Jk). Many of the antibodies fall to undetectable levels in the blood over time, so an anamnestic response to further antigen exposure may occur. Strategies to prevent alloimmunization include the use of leukodepleting filters, racially concordant donor–recipient matches, and extended red cell phenotyping. Antigen-negative blood is cryopreserved and stored through the American Red Cross Rare Donor Registry.
in cells containing hemoglobin SS, these cells have increased viscosity. Increasing the hematocrit by simple transfusion can worsen oxygen delivery to the tissues by raising blood viscosity. In general, anemia below the patient’s baseline level can be treated by simple transfusion. Red cell exchange transfusion is offered if rapid correction is needed in a patient with borderline cardiac function or volume overload, or if management of stroke or acute chest syndrome is desired (9). Although perioperative morbidity and mortality rates in sickle cell disease are improving, the potential for serious complications remains. Standard practice is to offer red cell transfusion to raise the hemoglobin to 10 g per dL with or without reducing hemoglobin SS levels to 30% for major surgical or ophthalmologic procedures. In addition to the usual risks of blood transfusion, these patients have problems with hyperviscosity and iron overload. Alloimmunization rates in patients with sickle cell disease are higher than those in the general population, approaching 20% in most series (Table 16-3). Historically, the most common antigens are Rhesus (Rh), Kell (K), Duffy (Fy), and Kidd (Jk). Many of the antibodies fall to undetectable levels in the blood over time, so an anamnestic response to further antigen exposure may occur. Strategies to prevent alloimmunization include the use of leukodepleting filters, racially concordant donor–recipient matches, and extended red cell phenotyping. Antigen-negative blood is cryopreserved and stored through the American Red Cross Rare Donor Registry.
TABLE 16-3 Alloimmunization Frequency in Sickle Cell Anemia Compared with Other Chronic Anemias. | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Thalassemia
The thalassemias are a heterogeneous group of genetic disorders of hemoglobin synthesis affecting production of alpha- or beta-globin. Clinically the alpha- and beta-thalassemias can be divided into more severe (major, intermedia,) and less severe (minor) disease. Hemolysis is associated with gall bladder disease, splenic dysfunction, hypersplenism, and total-body iron overload from chronic transfusion. Splenectomy is indicated in the management of chronically transfused patients in order to increase red cell survival and decrease transfusion requirements. Thalassemia is associated with a high risk of postsplenectomy infection and overwhelming sepsis (10).
Polycythemia
As the hemoglobin and red cell mass increase more than two standard deviations above the mean value for age, blood viscosity also increases. The most common causes of polycythemia are associated with increased erythropoietin production, either because of chronic hypoxia (cardiac or lung disease) or altered oxygen-carrying capacity of the hemoglobin within the red cells (hemoglobin M). One consideration in the decision to perform corrective surgery in patients with congenital heart disease is the fact that increasing polycythemia and hyperviscosity can result in thrombotic and ischemic events. Alterations in the erythropoietin receptor or signaling pathway have also been described. Excess erythropoietin production has been reported with Wilms tumor. Rarely, increased red cell mass occurs with normal erythropoietin levels: polycythemia vera is a myeloproliferative disorder that is treated with chemotherapy to lower red cell mass and prevent the complications of hyperviscosity. Phlebotomy has also been used to lower hemoglobin levels.
PLATELETS, VON WILLEBRAND FACTOR, AND FIBRINOGEN
Platelets participate in hemostasis by forming a platelet plug at the site of blood vessel injury and facilitating fibrin clot formation on their phospholipid membrane surface. Platelets circulate as nonthrombogenic disks, but upon exposure to various agonists released in response to vascular injury, they become activated, aggregate, and adhere to the site of disrupted endothelium. During platelet activation, platelet granules release their contents at the membrane surface and into the extracellular environment, causing activation and recruitment of additional platelets to the
area. Released clotting factors assist in the formation of a localized fibrin clot. Deficient granules or defective degranulation have been described in some patients with qualitative platelet disorders. A number of platelet membrane glycoproteins serve as receptors for adhesive polymeric proteins (11). Defective or deficient glycoproteins have been described in patients with rare, qualitative, congenital defects of platelet function. von Willebrand factor (vWF) binds the platelet glycoprotein Ib–glycoprotein IX complex and the subendothelium, allowing platelet adhesion to the vessel wall. The platelet plug continues to grow by linkage of platelets and fibrinogen through the glycoprotein IIb/IIIa receptors.
area. Released clotting factors assist in the formation of a localized fibrin clot. Deficient granules or defective degranulation have been described in some patients with qualitative platelet disorders. A number of platelet membrane glycoproteins serve as receptors for adhesive polymeric proteins (11). Defective or deficient glycoproteins have been described in patients with rare, qualitative, congenital defects of platelet function. von Willebrand factor (vWF) binds the platelet glycoprotein Ib–glycoprotein IX complex and the subendothelium, allowing platelet adhesion to the vessel wall. The platelet plug continues to grow by linkage of platelets and fibrinogen through the glycoprotein IIb/IIIa receptors.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


