•Periodic fever syndromes (PFS):
– Familial Mediterranean fever (FMF)
– Tumor necrosis factor receptor-associated periodic syndrome (TRAPS)
– Hyperimmunoglobulinemia D syndrome (HIDS)
– Periodic fever with aphthous stomatitis, pharyngitis and adenitis syndrome (PFAPA)
•Cryopyrin-associated periodic syndromes (CAPS):
– Familial cold autoinflammatory syndrome (FCAS)
– Muckle–Wells syndrome (MWS)
– Neonatal onset multisystemic inflammatory disorder (NOMID)
•CAPS related syndromes:
– Deficiency of the interleukin-1-receptor antagonist syndrome (DIRA)
– Deficiency of interleukin 36-receptor antagonist syndrome (DITRA)
•Other autoinflammatory syndromes:
– Pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA)
– NOD2-Associated pediatric granulomatous arthritis (PGA):Blau syndrome/early
– Onset sarcoidosis
– Majeed syndrome
– Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome
– Beçet syndrome
– Schnizltler syndrome
– Synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome
– Chronic recurrent multifocal osteomyelitis (CRMO) sporadic form
– Systemic juvenile idiopathic arthritis (Still disease) and adult onset
– Pyoderma gangrenosum, acne and suppurative hidradenitis (PASH)
– Possible autoinflammatory diseases:
– Inflammatory bowel disease
– Sweet syndrome, pyoderma gangrenosum and other neutrophilic dermatoses
Table 43.2
Classification of autoinflammatory diseases with dermatologic cutaneous findings
Disease | Defective gene |
|---|---|
Hereditary recurrent fevers | |
Familial Mediterranean fever | Pyrin (marenostrin) |
Mevalonate kinase deficiency (hyper IgD) syndrome | Mevalonate kinase (MVK) |
TNF receptor-associated periodic syndrome (TRAPS) | TNFRSF1A |
Cryopyrin associated periodic syndromes (CAPS) | |
CINCA/NOMID syndrome | NLRP3 |
Muckle–Wells syndrome | NLRP3 |
Familial cold autoinflammatory syndrome (FACS) | NLRP3 |
NLRP12-associated periodic fever (NAPS) | NLRP12 |
Pyogenic diseases | |
Deficiency of IL-1 receptor antagonist (DIRA) | IL1RN |
Deficiency of IL-36 receptor antagonist (DITRA) | IL36RN |
Pyogenic arthritis, pyoderma, acne (PAPA) syndrome | PSTPIP1 |
Majeed syndrome | LPIN2 |
Granulomatous and histiocytic diseases | |
Blau syndrome | NOD2 (CARD15) |
H syndrome | SCL29A3 |
Proteasome instability syndromes | |
CANDLE syndrome | PSMB8, others |
JMP syndrome | PSMB8 |
Nakajo–Nishimura syndrome | PSMB8 |
Others | |
Pityriasis rubra pilaris | CARD14 |
Disseminated superficial actinic porokeratosis | MVK |
Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation | PLCG2 (Phospholipase C, gamma 2) |
Hereditary Recurrent Fevers (Urticarial and Vasculitic Fevers)
Urticarial and erythematous eruptions are the cutaneous hallmark of the hereditary recurrent fevers
Cold-induced urticaria in the newborn is very suspicious of hereditary recurrent fevers
Amyloidosis may be an end-stage complication
This group encompasses AISs coursing with recurrent fevers and erythema. They present clinically with recurrent episodes of fever and erythematous swellings that can range from discrete urticarial wheals to large erythematous macules; the lesions can occur daily or at variable intervals from days to weeks. Several clinical manifestations of systemic inflammation accompany the eruption.
Familial Mediterranean Fever
Familial Mediterranean fever (FMF) is due to mutations in the MEFV gene, codifying pyrin or marenostrin
Erysipeloid plaques are the most characteristic cutaneous lesions
Colchicine may control the disease
FMF is the paradigm of the periodic fever syndromes. It is an autosomal recessive (AR) disorder due to mutations in the gene MEFV (MEditerranean FeVer), which encodes a protein named pyrin or marenostrin [2, 3]. Pyrin is a main regulatory component of the NLRP3 inflammasome, a large protein complex that takes part in inflammation signaling pathways such as the IL-1β pathway [4].
Clinical Manifestations
FMF patients present episodic attacks of fever that typically last less than 72 h, abdominal pain, pleurisy, mono or oligoarticular arthritis, and/or arthralgias [5]. Attacks are self-limited and between them the patient remains asymptomatic. The interval between attacks is variable. Exercise, emotional stress, exposure to extreme temperatures, and hormonal changes can trigger the attacks [5]. Amyloidosis is a severe complication that could appear in untreated patients. Erysipeloid-like lesions, which appear in 15–20 % of children, are the characteristic cutaneous manifestation of FMF [6–8]. They usually appear in the lower extremities as well-circumscribed edematous and erythematous plaques. Histopathology shows a predominantly neutrophilic infiltrate with nuclear dust [9]. Purpuric lesions on the face, trunk, and extremities have also been reported in children.
Treatment
Colchicine is the treatment of choice; it reduces the frequency, intensity, and the duration of the attacks [10]. In patients who do not respond to colchicine, it is important to verify the compliance with therapy. Intravenous colchicine could be tried as some of these not-responders do respond to this regime. There are isolated reported cases of truly colchicine-resistant patients that responded to thalidomide, to anti-TNFα agents such as etanercept and infliximab, and to the recombinant IL-1 receptor antagonist anakinra [11, 12].
Cryopyrin-Associated Periodic Syndromes
Cryopyrin-associated periodic syndromes (CAPS), also called cryopyrinopathies, are a group of three overlapping autosomal dominant syndromes, which are due to self-activating mutations in the NLRP3 gene (also called CIAS1). NLRP3 product is the protein called cryopyrin, which is part of a multiprotein inflammasome complex (the NLRP3 inflammasome), which in response to certain stimuli activates the caspase 1 cascade that leads to production of pro-inflammatory cytokines, mainly IL-1β and IL-18 [13].
Clinical Manifestations
Neonatal urticaria is the earliest sign of the disease
Peculiar cherubic face is seen
Arthropathy and CNS are often involved
CAPS include familial cold autoinflammatory syndrome (FCAS, or familial cold urticaria), Muckle–Wells syndrome (MWS, urticaria-deafness-amyloidosis syndrome), and neonatal onset multisystemic inflammatory disorder (NOMID, also named as chronic infantile neurological cutaneous and articular (CINCA) syndrome). NOMID is the most severe form of CAPS. Symptoms appear shortly after birth, usually before 6 months of age. The triad of fevers, skin urticarial rash, severe arthropathy, and central nervous system disorders characterizes NOMID [14, 15] (Fig. 43.1). Patients with NOMID show abnormal facial features, including flattening of the nasal bridge, macrocephaly, frontal bossing, and protruding eyes [16]. Neurological manifestations develop with time, and include chronic aseptic meningitis, cerebral atrophy, sensorineural hearing loss, and developmental delay. Eye manifestations, such as anterior uveitis, papilledema, or blindness, also occur in NOMID [17]. About 50 % of patients have severe arthropathy before 12 months of age. Secondary amyloidosis may appear due to chronic inflammation [1]. An erythematous skin eruption resembling urticaria is usually present in the neonatal period or may appear before 6 months of age. The rash persists during the whole life of patients. On histopathology, superficial and deep perivascular infiltrates are seen composed mainly of neutrophils, lymphocytes, and some eosinophils [18].
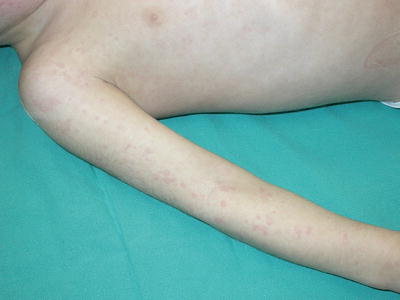
Fig. 43.1
Urticarial rash in NOMID syndrome
Pyogenic Disorders
Pustules on an erythematous basis in a newborn with fevers are the main presentation
Osteoarticular symptoms are frequent
Histopathology resembles pustular psoriasis in many cases
The deficiencies of the interleukin-1-receptor antagonist syndrome (DIRA) and of interleukin-36-receptor antagonist syndrome (DITRA) are the paradigm of pustular eruptions associated with periodic fever. Other diseases in this group include pyogenic arthritis, pyoderma, acne (PAPA) syndrome, and Majeed syndrome. In all of them, the main cutaneous lesions are pustular eruptions, which show marked neutrophil infiltration with prominent intraepidermal neutrophil accumulations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


