Ambiguous Genitalia in the Newborn and Disorders of Sex Development
Ingrid Holm
Although most clinicians rarely see an infant with ambiguous genitalia at birth, the need to assess the situation as quickly as possible makes this subject essential. Any deviation from the normal appearance of male or female genitalia should prompt investigation, since apparent but incomplete male or female external genitals may be associated with the gonads and genotype of the opposite sex. Even a slight doubt that arises during the initial examination of the newborn should be pursued systematically to prevent the possibility of later confusion. Bilateral cryptorchidism, unilateral cryptorchidism with incomplete scrotal fusion or hypospadias, labial fusion, or clitoromegaly requires evaluation.
Terminology
The Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology organized an International Consensus Conference on Intersex and published a consensus statement (1) proposing the term “disorders of sex development” (DSD) to define “congenital conditions in which development of chromosomal, gonadal, or anatomic sex is atypical.” A subset of children with DSD have disorders that present at birth with ambiguous genitalia. The DSDs are classified as sex chromosome DSD, 46,XY DSD (formerly “male pseudohermaphrodite” or “undervirilized male”), and 46,XX DSD (“formerly female pseudohermaphrodite” or “overvirilized female”).
Determining Sex Assignment
Abnormal sexual differentiation presents medical and psychological issues that need to be addressed in an urgent fashion. Two of these major issues need to be immediately addressed: the relationship of the sexual ambiguity to a possible life-threatening disease and the sex of rearing. In the delivery room, it is critical not to make a gender assignment but to postpone that determination until the necessary data have been collected. Clearly, most parents will react with dismay and anxiety. It is important to reassure the parents that they have a healthy baby but that the development of the external genitals is incomplete and tests are necessary to determine the sex. They should be reassured that tests will be performed that will show the cause of the problem and identify the baby as a girl or a boy and that a definite answer should be possible within a few days, or at most in 1 or 2 weeks. The possibility of a DSD does not need to be raised initially. Speculation about the possible assignment of sex could be psychologically damaging to the parents. The child should be referred to by all caretakers as the baby rather than as a boy or girl. Parents should be encouraged to tell other family members and friends, when they ask about the sex of the baby, that the baby is sick, and to delay sending out birth announcements until the gender options have been considered. The physician should examine the baby in the presence of the parents, explain the common genital anlage for boys and girls, and educate the family about normal sexual development. Once the diagnosis is made, the natural history, prognosis, and therapeutic options should be discussed with the parents. Full disclosure and communication are essential. Decisions regarding sex assignment must be made with the parents, taking into account their cultural and religious beliefs and level of understanding. Parents should be made aware of recent controversies in this area of sex assignment and provided with educational information and resources. Once the sex of rearing has been determined, the physician should help the family put aside issues of sexual ambiguity. Parents should be encouraged to use the names previously selected for a boy or girl. Names that are definitely male or female help the family see the child unequivocally as belonging to that sex. As long as parental attitudes toward the child’s sex remain unequivocal, the child usually assumes his or her gender role without difficulty, regardless of the genotype.
Although a diagnosis of the patient’s condition requires knowledge of the genotype (karyotype), sex assignment is generally based on other criteria as well. The main issues are the potential for an unambiguous appearance, normal sexual functioning, and fertility. The individual with a 46,XX DSD has normal ovaries and uterus, is potentially capable of bearing children, and thus should usually be given a female gender assignment. In general, the individual with a 46,XY DSD should be raised as a male, unless there is complete androgen resistance, the genitalia are almost completely or completely feminized, or the family makes an informed decision to raise the child as a female. In sex chromosome DSDs in which fertility is not possible, the decision regarding sex assignment should be based, in part, on the potential for reconstructive surgery. In general, surgical techniques are more suited to reduction of the size of the phallus and, later, to the creation of a vagina than to the construction of a normal male phallus.
In the evaluation of children with ambiguous genitalia, it is critical to provide a multidisciplinary approach. Psychiatrists and social workers should be included as sex assignment is being determined, and in the long-term management. The decision to make a sex assignment must be carefully considered with the knowledge that there are those who advocate making no sex assignment and allowing the child to choose for himself or herself, when old enough to do so. This approach is advocated by intersex societies (Intersex Society of North America). Thus, the family must be made aware of all options and allowed to choose, even if the medical team has come to a consensus as to the best sex assignment. (See reference [2] for a review of the psychological ramifications of sex assignment in children with ambiguous genitalia.)
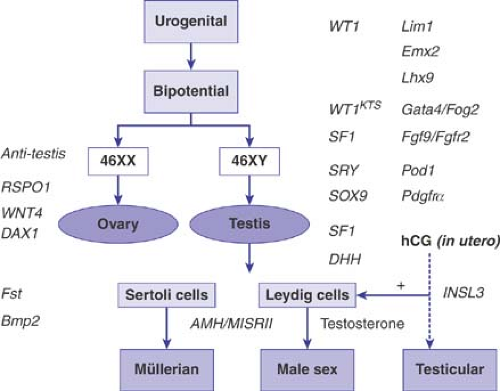 Figure 3-1. Summary of genes involved in fetal sex development. Genes denoted in lower case refer to those which, when disrupted, lead to sex reversal in the mouse only. Hcg = human chorionic gonadotropin. (From Hughes IA, Nihoul-Fékété C, Thomas B, et al. Consequences of the ESPE/LWPES Guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab 2007;21(3):351–365.) |
Review of Embryogenesis
Table 3-1 Mutations in Genes Involved in Sex Determination and Development, and Associated with Intersex Anomalies | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
The bipotential gonads develop prior to the fifth to sixth week of gestation. Steroidogenic factor 1 (SF1) and Wilms tumor 1 (WT1) are critical for the development of the genital ridge into the bipotential gonad prior to sexual differentiation. Mutations in SF1 and WT1 are associated with abnormal gonadal development, resulting in 46,XY sex reversal or ambiguous genitalia. Other genes proposed to be involved in gonadal development include LIM1 and EMX2, although no mutations responsible for sex reversal have been found in these genes (Fig. 3-1 and Table 3-1) (see references [3–5] for a review of genes involved in sex
determination and differentiation). The bipotential gonads differentiate into either testes or ovaries. In general, maleness is imposed upon the innate tendency of the fetal gonads to develop along female lines. There are three major components to sex determination and differentiation: chromosomal sex, gonadal sex, and phenotypic sex. Chromosomal sex refers to the karyotype, 46,XY or 46,XX, which under normal circumstances determines whether the individual is male or female. More specifically, the chromosomal sex refers to genes important in sex determination (i.e., the events that lead to the differentiation of the gonads into either an ovary or testis) (5). The first critical gene in the cascade of genes involved in sex determination is the sex-determining region of the Y chromosome (SRY), which is located on the short arm of the Y chromosome (Yp) just centromeric to the pseudoautosomal region (the region where the X and Y chromosomes pair during meiosis). SRY is required for differentiation of the gonad into a testis (6,7). If it is absent or abnormal, the gonad differentiates into an ovary. The importance of SRY in sex determination is demonstrated by 46,XX males who have SRY present (7) and 46,XY females with mutations or deletions in SRY (6,8). The SRY protein activates transcription of genes important in sex determination (6,7,8,9,10). Defects in genes activated by SRY are postulated to be responsible for sex reversal in 46,XX males who have no Y sequences detectable (11,12), including no SRY, and in 46,XY females with normal SRY (13,14).
determination and differentiation). The bipotential gonads differentiate into either testes or ovaries. In general, maleness is imposed upon the innate tendency of the fetal gonads to develop along female lines. There are three major components to sex determination and differentiation: chromosomal sex, gonadal sex, and phenotypic sex. Chromosomal sex refers to the karyotype, 46,XY or 46,XX, which under normal circumstances determines whether the individual is male or female. More specifically, the chromosomal sex refers to genes important in sex determination (i.e., the events that lead to the differentiation of the gonads into either an ovary or testis) (5). The first critical gene in the cascade of genes involved in sex determination is the sex-determining region of the Y chromosome (SRY), which is located on the short arm of the Y chromosome (Yp) just centromeric to the pseudoautosomal region (the region where the X and Y chromosomes pair during meiosis). SRY is required for differentiation of the gonad into a testis (6,7). If it is absent or abnormal, the gonad differentiates into an ovary. The importance of SRY in sex determination is demonstrated by 46,XX males who have SRY present (7) and 46,XY females with mutations or deletions in SRY (6,8). The SRY protein activates transcription of genes important in sex determination (6,7,8,9,10). Defects in genes activated by SRY are postulated to be responsible for sex reversal in 46,XX males who have no Y sequences detectable (11,12), including no SRY, and in 46,XY females with normal SRY (13,14).
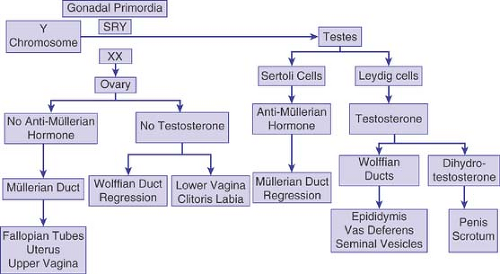 Figure 3-2. Male and female gonadal differentiation. Gonadal primordia, under the influence of the sex-determining region (SRY), become a testis; the presence of antimüllerian hormone (AMH) and testosterone causes the regression of müllerian ducts and the formation of male internal and external genitalia. In the absence of SRY, an ovary differentiates, and in the absence of AMH and testosterone, female internal and external genitalia are formed. |
Sex reversal occurs in patients with abnormalities in SOX9, a gene that functions downstream of SRY (9,10). SOX9 encodes a transcription factor related to SRY and is involved in testicular development. Mutations in SOX9 disrupt male development and cause 46,XY sex reversal as well as camptomelic dysplasia (15,16,17). SF1 and WT1 interact and are also involved in testicular development. The dosage-sensitive sex reversal gene on the X chromosome (DAX1) antagonizes this interaction and inhibits the gonad from developing into a testis (3,18). In addition, mapping of the Y chromosome has identified regions involved in spermatogenesis (19,20,21), although the nature of these genes is unclear.
The presence of two X chromosomes appears to be important for development of a normal functioning ovary; 46,XX and 45,X fetuses initially have oocytes. However, in the 45,X fetus, oocyte atresia is accelerated in the second half of intrauterine life and in the prepubertal years. This suggests that two copies of genes on the X chromosome are necessary for oocyte maintenance. Mapping of the X chromosome has identified regions of the X chromosome responsible for short stature, some of the stigmata of Turner syndrome, and ovarian function. Duplications in DAX1 in 46,XY individuals leads to sex reversal. It now appears that DAX1 functions by inhibiting the gonad from developing into a testis, and is not an ovary-determining gene (22). WNT4 (23), RSPO1, and FOXL2 (24,25) are also thought to play a role in the development of the ovary. Overall, however, the genes involved in the development and maintenance of the ovary are still largely unknown.
The gonadal sex is established under the influence of the chromosomal sex. The first sign of gonadal differentiation occurs in the male with the appearance of Sertoli cells at 6 to 7 weeks’ gestation (26); Leydig cells appear at about 8 weeks. In the female at this stage, the only sign of ovarian differentiation is the absence of Sertoli and Leydig cells. At 9 weeks’ gestation primordial germ cells begin to differentiate into oogonia, and by 12 to 13 weeks, the ovary begins to develop (26). In contrast to the male, in whom testicular cords form in the absence of germ cells, the ovary does not develop if germ cells are not present.
The end result of gonadal differentiation (testis or ovary) determines the hormone that will be secreted to lead to the phenotypic sex. Sex differentiation is the process by which hormone secretion, and the response of end organs to these hormones, results in the phenotypic sex (Fig. 3-2). Testosterone and antimüllerian hormone (AMH), secreted from the Leydig cells and the Sertoli cells of the testes, respectively, induce differentiation of the genital primordia into the male phenotype. In the absence of a functioning testis, whether or not an ovary is present, the genital primordia will differentiate into the female phenotype. However, although the presence of a functioning testis is necessary, it is not sufficient for the development of a male phenotype. Normal androgen metabolism is also required, including normal androgen receptors and 5α-reductase activity.
The internal genitalia are derived from the müllerian ducts in the female and the wolffian ducts in the male (Fig. 3-3). The first phenotypic sign of differentiation of the male internal genitalia is the regression of müllerian ducts at 8 weeks of gestation, mediated by AMH (26). This hormone, a high-molecular-weight glycoprotein secreted from the Sertoli cells, prevents the differentiation of the müllerian duct structures (the fallopian tubes, uterus, and upper vagina); the müllerian duct structures are almost completely absent by 10 weeks. Testosterone secreted from the Leydig cells is responsible for the differentiation of the wolffian ducts (the vas deferens, seminal vesicles, and epididymis). High concentrations of AMH and testosterone are required locally around the müllerian and wolffian ducts, respectively, and androgen receptors must be functional, for differentiation of the male internal genitalia to occur. In the female, or in the absence of testicular tissue–secreting AMH and testosterone locally, müllerian duct differentiation and wolffian duct regression occur. This explains why 46,XX DSD individuals do not have testes and develop female internal genital structures.
External genital development occurs between the 8th and 12th weeks of gestation and in the male does not require high local concentrations of testosterone. Normal differentiation of
the male external genitalia requires high circulating levels of testosterone, the conversion of testosterone to dihydrotestosterone (DHT) by 5α-reductase in the target organs, and functional androgen receptors. In the male, under the influence of DHT, the urogenital sinus gives rise to the prostate, the genital tubercle forms the glans penis, the labiourethral folds form the urethra and ventral shaft of the penis, and the labioscrotal folds fuse to form the scrotum. In the female, or in the absence of testicular tissue secreting bioactive testosterone, functioning androgen receptors, or 5α-reductase, the genital tubercle forms the clitoris, the labiourethral folds form the labia minora, and the labioscrotal folds form the labia majora.
the male external genitalia requires high circulating levels of testosterone, the conversion of testosterone to dihydrotestosterone (DHT) by 5α-reductase in the target organs, and functional androgen receptors. In the male, under the influence of DHT, the urogenital sinus gives rise to the prostate, the genital tubercle forms the glans penis, the labiourethral folds form the urethra and ventral shaft of the penis, and the labioscrotal folds fuse to form the scrotum. In the female, or in the absence of testicular tissue secreting bioactive testosterone, functioning androgen receptors, or 5α-reductase, the genital tubercle forms the clitoris, the labiourethral folds form the labia minora, and the labioscrotal folds form the labia majora.
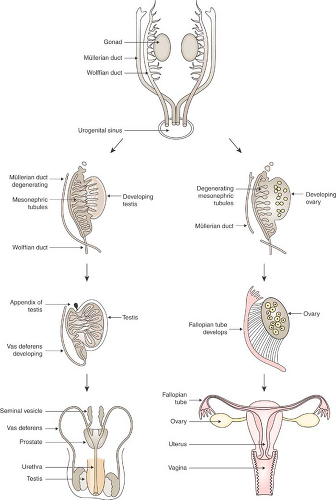 Figure 3-3. Differentiation of the wolffian and müllerian ducts. (From Warne GL, Kanumakala S. Molecular endocrinology of sex differentiation. Semin Reprod Med 2002;20:174; with permission of Thieme Medical Publishers.) |
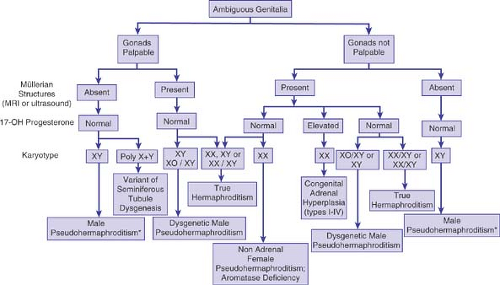 Figure 3-4. Steps in the diagnosis of intersexuality in infancy and childhood. (From Grumbach MM, Conte FA. Disorders of sex differentiation. In: Wilson JD, Foster DW, Kronenberg HM, et al., eds. Williams textbook of endocrinology. Philadelphia: WB Saunders, 1998; with permission of W.B. Saunders Company.) |
Thus, from this brief review of embryogenesis, it is clear that a defect in any one of many steps along the pathway of sex determination and differentiation can result in ambiguous genitalia.
Assessment of the Neonate
The initial evaluation of the newborn with ambiguous genitalia includes a careful history, physical examination, karyotype determination, serum hormone analyses, and radiographic studies (Fig. 3-4). The direction of further studies, including the performance of a human chorionic gonadotropin (HCG) stimulation test, more extensive hormone analyses, DNA studies in a search for SRY or mutations in other genes, laparoscopy, or gonadal biopsy, depends on the results of the initial evaluation.
History
A careful history should be obtained from the parents, particular attention being given to the following points:
Family history (Table 3-2)
Other family members, especially siblings, with congenital adrenal hyperplasia (CAH).
A history of early neonatal death. The diagnosis of CAH may be missed in males, since there are often no physical signs at birth except, occasionally, increased scrotal rugae, pigmentation, or a large phallus. Thus, a brother may have died in early infancy of vomiting and dehydration, not recognized as secondary to adrenal insufficiency.
Consanguinity, which makes an autosomal recessive disorder, such as CAH or 5α-reductase deficiency, more likely.
Aunts or other relatives with amenorrhea and/or infertility, which may suggest a 46,XY DSD.
Maternal history
Maternal ingestion of drugs during pregnancy (particularly androgens or synthetic progestational agents) as well as antiandrogens (such as spironolactone).
Maternal exposure to chemical endocrine disrupters in the environment (27).
Maternal history of virilization or CAH.
Physical Examination
Table 3-2 Patterns of Inheritance: Ambiguous Genitalia | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
The physical examination of the infant starts with a general examination and a search for the stigmata of a malformation syndrome (e.g., intrauterine growth retardation, dysmorphic
features, and abnormal body proportions). If a malformation syndrome is identified, further hormonal evaluation may not be necessary (e.g., if the suspected diagnosis is trisomy 13). Other signs to look for on the general examination include hypertension, areolar hyperpigmentation, and signs of dehydration, all suggestive of CAH.
features, and abnormal body proportions). If a malformation syndrome is identified, further hormonal evaluation may not be necessary (e.g., if the suspected diagnosis is trisomy 13). Other signs to look for on the general examination include hypertension, areolar hyperpigmentation, and signs of dehydration, all suggestive of CAH.
The genitalia should be carefully examined. The number of gonads and the size, symmetry, and position are crucial. A palpable gonad below the inguinal canal is almost always a testis and, if present, generally rules out the diagnosis of 46,XX DSD. An undescended gonad could be a testis, ovary, or ovotestis. Asymmetric labioscrotal folds with a unilateral gonad make mixed gonadal dysgenesis more likely. The infant should be examined for the presence of a hernia, as it may contain a uterus, ovary, or testis. Precise measurements of the phallus should be made, including the stretch penile length (along the dorsum of the stretched phallus from the pubic ramus to the tip of the glans) and the midshaft diameter. The mean stretched penile lengths in normal males are as follows (±1 standard deviation):
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


