Disorders of Granulocytes
R. Alexander Blackwood and Lawrence A. Boxer
White blood cells, or leukocytes, are part of the innate immune system. They are one of the body’s major defenses in preventing and combating infection. The most common types of leukocytes are lymphocytes, monocytes, and granulocytes. This chapter will deal primarily with granulocytes. These cells are derived from stem cells in the bone marrow and have diverse functions. The most common of the leucocytes is the neutrophil. Deficiency of neutrophils, called neutropenia, is one of the most common hematologic abnormalities during childhood and when severe can result in life-threatening infection.
NEUTROPENIA
The absolute neutrophil count and the relative proportions of neutrophils to lymphocytes vary with age. At birth, neutrophils predominate but rapidly decrease during the first few days of life. During infancy, neutrophils comprise approximately 20% to 30% of the circulating white blood cell population. By 5 years of age, neutrophil and lymphocyte counts are equal, and by puberty, adult proportions of approximately 70% are reached.
Neutropenia is defined by an absolute decrease in the absolute number of circulating mature (segmented) and band forms of neutrophils, which can be determined by calculating the absolute neutrophil count (ANC). The ANC is calculated by multiplying the total white blood cells (WBC) obtained from the complete blood count (CBC) by the combined percentage of segmented neutrophils and bands from the differential. The normal resting ANC in the general population ranges between 1500 and 8000 cells/cm3 for Caucasian children over 6 years of age, while 30% of African American children have an ANC as low as 1000 cells/μl. Based on the ANC, neutropenia can be classified as mild (ANC between 1000–1500 cells/μl), moderate (ANC between 500–1000 cells/μl), or severe (ANC < 500 cells/μl). Neutropenia is associated with an increased chance of developing infections. 
CLASSIFICATION OF PRIMARY NEUTROPENIAS
The inherited disorders of bone marrow production, characterized by selective loss of neutrophil production without accompanying congenital anomalies, are diagnosed by clinical and laboratory features and by genetic testing (Table 441-1).1
DISORDERS OF GRANULOPOIESIS
Reticular dysgenesis (RD) is a very rare form of severe combined immunodeficiency characterized by agammaglobulinemia, lymphopenia, and neutropenia.2-4 Patients with RD usually die shortly after birth from overwhelming sepsis. Bone marrow transplantation is the only available treatment. Examination of the bone marrow from patients with RD reveals absent myeloid precursors with normal erythroid and platelet differentiation. RD is thought to arise from a defect in a common stem cell that affects maturation of myeloid and lymphoid cell lines. 
Cyclic neutropenia is an autosomal dominant disorder resulting from a mutation in the ELA2 gene encoding for neutrophil elastase.5-8 Peripheral blood neutrophil numbers oscillate with a 21 ± 3 day periodicity, with the nadirs often falling below 200 cells/μl. During the periods of neutropenia, patients often have episodes of fever, malaise, aphthous stomatitis, cervical lymphadenopathy, and gastroenteritis. While episodes may be asymptomatic, life-threatening infections with Clostridia perfringens and gram-negative organisms are a risk. The diagnosis can be made by monitoring peripheral neutrophil counts three times a week over a 6- to 8-week span. The diagnosis can be confirmed by sequencing the ELA2 gene.7,9 While management is predominantly symptomatic, aggressive use of antibiotics, including coverage for both gram-negative and anaerobic organisms, during infections may be required. Improvement in the neutrophil count occurs with daily granulocyte colony-stimulating factor (G-CSF). Unlike patients with severe congenital neutropenia (SCN), these patients are not at an increased risk for hematologic malignancies.
Table 441-1. Classification of Congenital Neutropenia Syndromes
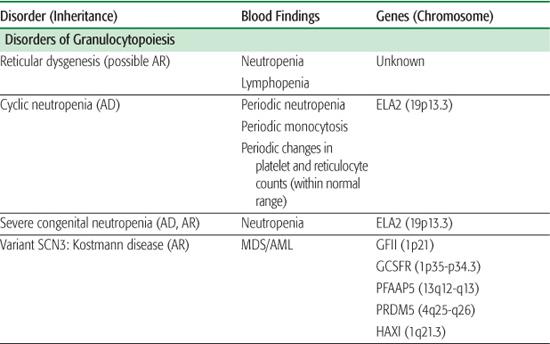
Chronic benign neutropenia represents a group of disorders that are characterized by absolute neutrophil counts (ANCs) persistently less than 1500 cells/μl. Patients with this disorder are not at an increased risk of developing severe infections but may present with mild to moderate infections of the skin and mucous membrane, including oral ulcers, chronic gingivitis, and chronic periodontitis. Inheritance is autosomal dominant or sporadic, and the management consists of good skin and dental care and expeditious use of antibiotics for infections.
Severe congenital neutropenia and Kostmann disease10,11 are a group of genetic disorders characterized by severe neutropenia, with ANCs persistently below 0.2 × 103 cells/μl and with associated monocytosis and eosinophilia. Affected patients suffer from severe life-threatening bacterial infections beginning in the first few months of life. Examination of the bone marrow reveals an arrest in myeloid differentiation at the promyelocyte or myelocyte stage of development. Approximately 60% of cases of SCN arise from a mutation (autosomal dominant or sporadic) in the ELA2 gene, resulting in an increase in apoptosis of neutrophil precursors in the bone marrow. 
A majority of patients respond to G-CSF, resulting in an increase in peripheral neutrophil counts and reduced rates and severity of infections.15 While their prognosis is greatly improved, they are still at risk for infection, and they have up to a 40% risk of developing myelodysplasia (MDS) or acute myelogenous leukemia (AML). 
MULTIFACETED SYNDROMES ACCOMPANIED BY NEUTROPENIA 
 DISORDERS OF RIBOSOMAL DYSFUNCTION
DISORDERS OF RIBOSOMAL DYSFUNCTION
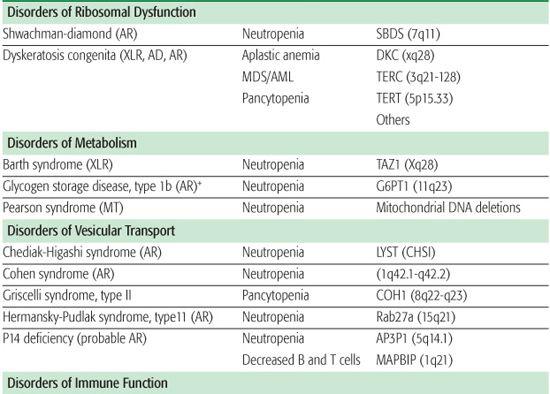
Shwachman-Diamond syndrome (SDS) is an autosomal recessive disorder characterized by neutropenia, recurrent infections, exocrine pancreatic insufficiency, short stature, and metaphyseal dysplasia.17 Metaphyseal chondrodysplasia, associated with dwarfism, occurs in approximately 50% of patients, but other skeletal anomalies include rib cage deformities, osteopenia, clinodactyly, kyphosis, supernumerary metatarsals, coxa vara deformity, and syndactyly. SDS usually presents in early infancy due to pancreatic insufficiency with steator-rhea, weight loss, and failure to thrive. While anemia and thrombocytopenia can occur, neutropenia is the most common hematologic manifestation of SDS. Approximately 90% of patients with SDS have an inactivating mutation in the SBDS gene (located on chromosome 7q11), which codes for a protein involved in rRNA maturation.18,19 These patients also have a propensity for monosomy 7–related hemato-logic disorders, including MDS or AML.
Management of SDS must include the replacement of pancreatic enzymes. G-CSF will raise the absolute neutrophil count into the normal range but should be reserved for persistent neutropenia associated with recurrent bacterial infection. MDS and AML are generally preceded by cytogenetic changes in the bone marrow. Bone marrow transplantation may be curative, but its precise role has not been completely defined.17
Dyskeratosis congenita (DC) is a rare congenital syndrome in which progressive bone marrow failure is associated with the triad of reticulated skin hyperpigmentation, nail dystrophy, and oral leukoplakia.20-22
 DISORDERS OF METABOLISM
DISORDERS OF METABOLISM
Barth syndrome is a rare X-linked recessive neuromuscular and metabolic disorder characterized by cardiomyopathy, skeletal muscle weakness, and neutropenia and growth retardation. 
 DISORDERS OF VESICULAR TRANSPORT
DISORDERS OF VESICULAR TRANSPORT
Chediak-Higashi syndrome (CHS) is a rare autosomal recessive disorder characterized by partial oculocutaneous albinism in conjunction with neutrophil dysfunction.30 This disorder is frequently associated with recurrent infections; enterocolitis; peripheral neuropathy; easy bruis-ability with prolonged bleeding times; and hypopigmentation of the skin, hair, and eyes.
CHS is caused by a mutation in the lysosome trafficking regulatory gene (LYST) localized to chromosome 1 (q42-43) resulting in a defect in intracellular trafficking that affects vesicle formation and fusion.31,32 Neutrophils contain giant specific and azurophil granules exhibiting diminished bactericidal activity. Chemotactic activity is decreased, and intracellular killing is diminished due to a slow, inconsistent delivery of dilute amounts of hydrolytic enzymes from the giant granules into the phagosomes. 
Most patients exhibit an accelerated phase caused by a lymphohistiocytic proliferation in the reticuloendothelial systems, which intensifies the already existing neutropenia and leads to pancytopenia.33
Oculocutaneous albinism is prominent in chediak-Higashi syndrome (CHS) secondary to melanocyte dysfunction. Patients with this condition have skin that is usually fair, light blond or silvery gray hair, pale retinas, and translucent irises. 
CHD is often fatal before age 10, usually secondary to overwhelming infection. Those who survive longer are at increased risk of developing lymphomas. Early bone marrow transplantation is preferred therapy, especially if the patient is in the accelerated phase. 
 DISORDERS OF IMMUNE FUNCTION
DISORDERS OF IMMUNE FUNCTION
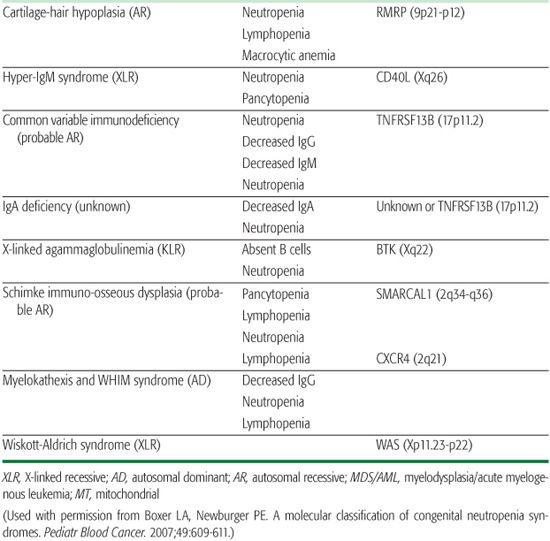
Cartilage-hair hypoplasia (CHH) is an autosomal recessive disorder characterized by short-limb dwarfism arising from metaphyseal dysplasia, fine hair, immunodeficiency, and increased incidence of cancer.40,41
Hyper-IgM syndrome is an immunodeficiency disorder with elevated IgM caused by at least three different genetic defects. The most common form of the disease, X-linked hyper-IgM syndrome (Xq26-27.2), arises from mutations in the gene for CD40 ligand and in 50% of patients is accompanied by neutropenia.42,43 The mechanism of neutropenia is not well understood. Myeloid precursors in the bone marrow express CD40 ligand, and bone marrow examination in these patients reveals maturation arrest at the promyelocyte-myelocyte stage. 
Wiskott-Aldrich syndrome is an X-linked genetic disorder with variable expression and in rare cases severe neutropenia.44 Those patients with neutropenia have cellular and humoral immunodeficiency, normal platelet numbers, and recurrent infections. The Wiskott-Aldrich syndrome protein (WASP) is located on Xp11.22-23 and codes for a protein involved in signal transduction between G-protein coupled receptors and the initiation of actin bundling. WASP mutations affect cellular functions such as growth, endocytosis, exocytosis, and cytokinesis. The bone marrow shows trilineage dysplasia with markedly reduced myelopoiesis. The neutropenia responds to G-CSF therapy. 
ACQUIRED NEUTROPENIAS OF MYELOID AND STEM CELLS
Neutropenia can develop as a result of a variety of conditions.48 Bone marrow failure with neutropenia, associated with anemia and thrombocytopenia, can occur in aplastic anemia, which is thought to arise from altered immune function or from marrow replacement (eg, malignancy). Vitamin B12, folate, and copper deficiency as well as starvation can lead to decreased marrow production of neutrophils. Antibodies directed against neutrophils and several drugs, including chemotherapeutic agents, can induce neutropenia. Overwhelming bacterial sepsis, some viruses, fungi, and protozoal infections can also cause neutropenia. This condition can arise during hemodialysis and leukapheresis. Many of the acquired neutropenias will respond to removal of the offending agent or treatment with recombinant G-CSF.
IMMUNE NEUTROPENIAS
Alloimmune neonatal neutropenia is a self-limited condition whereby maternal antineutrophil antibodies (IgG) cross the placenta, resulting in the destruction of the newborn’s neutrophils.49 The duration and extent of the neutropenia depends on maternal antibody production, but recovery is usually complete within 6 to 12 weeks. 
Autoimmune neutropenia of infancy is a relatively benign condition in which children transiently develop moderate to severe neutropenia secondary to the production of antineutrophil antibodies.50 While the absolute neutrophil counts often fall below 0.5 × 103 cells/μl, life-threatening infections rarely occur. Peripheral neutrophil counts respond to G-CSF. 
Autoimmune neutropenia arising from the production of antineutrophil antibodies can occur in patients with no underlying autoimmune disorder or can be associated with a variety of autoimmune conditions such as systemic lupus erythematosus, autoimmune hemolytic anemia, immune thrombocytopenia, or thyroiditis. Autoimmune neutropenia in children frequently is associated with immune deficiencies.51 Successful management of the underlying autoimmune disease will often result in normalization of peripheral neutrophil counts. In other instances, G-CSF may be required.
Drug-related neutropenia is common and can occur at any age, although more than 90% of cases occur in adults.52 Drug-induced neutropenia is not always predictable, but when it arises, it may occur by several different mechanisms, including immune-mediated toxic, idiosyncratic, and hypersensitivity reactions. In contrast, cytotoxic chemotherapy predictably affects proliferating myeloid cells by impairing neutrophil production. Therapy of the former causes of neutropenia requires cessation of the responsible drug. Treatment with G-CSF should be employed, but it may not always be effective. The neutropenia accompanying cytotoxic chemotherapy typically occurs 7 to 10 days after administering the drug. It is often accompanied by depressed cellular immunity, thereby predisposing patients to a much greater risk of infection than those disorders associated with the acute onset of isolated neutropenia.
Reticuloendothelial sequestration frequently results in mild to moderate neutropenia often accompanied by platelet and red cell trapping in the spleen, leading to thrombocytopenia and anemia. Splenectomy should be avoided if possible because of the increased risk of infection due to encapsulated bacterial organisms.48
Viral-related neutropenia occurs concurrently or shortly following a viral illness and is secondary to viral antibodies cross-reacting to neutrophil antigens. The neutropenia is usually mild, transient, and of minimal clinical significance. Conditions that result in complement activation, inflammatory disorders, hemodialysis, and extracorporeal membrane oxygenation (ECMO) can also cause neutropenia.53
NEUTROPHIL PRODUCTION AND KINETICS
Neutrophils originate from pluripotential stem cells in the bone marrow. Depending on environmental influences, these stem cells may give rise to the early progenitors of blood cells. These pluripotential stem cells give rise to more mature stem cells that are committed to either lymphoid or myeloid development. Myelopoiesis begins with about 106 stem cells in the bone marrow; these cells undergo both self-renewal and differentiation to produce all the individual types of blood cells. 
NEUTROPHIL FUNCTION
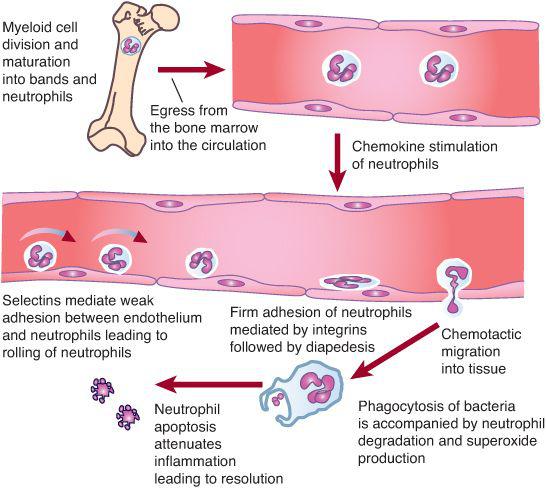
Neutrophils are the first line of defense against bacterial and fungal infections. About 1011 neutrophils per day are produced.30 Neutrophil responses are initiated as these cells pass through the postcapillary venules and detect low levels of chemokines and other substances released from sites of infection. During infection, the cellular detectors initiate subtle changes in the distribution and activity of surface molecules on both endothelial cells and neutrophils (Fig. 441-1). The initial associations lead to weak neutrophil adherence, as manifested by the rolling of the neutrophils on the endothelium. Rolling is characterized by neutrophil adhesion, which is made and broken, thereby allowing neutrophils to move hesitantly along the endothelial surface. 
When the neutrophil reaches the site of infection, it recognizes pathogens by means of Fc immunoglobulin, complement, and toll-like receptors. The neutrophil ingests the microbes that are opsonized (prepared for ingestion) by factors in human serum, which include immunoglobulin G (IgG) and C3, respectively. The opsonins facilitate phagocytosis of microbes into a closed vacuole called a phagosome. As phagocytosis is proceeding, two cellular responses essential for optimal microbicidal activity occur concomitantly—degranulation and activation of the nicotinamide-adenine dinucleotide phosphate (NADPH)–dependent oxidase. Fusion of the neutrophil granule membranes with the phagosome membrane results in the delivery of both components from the azurophil and specific granules. Concomitantly, the assembly and activation of NADPH oxidase at the phagosome membrane occurs. The multicomponent NADPH oxidase generates superoxide (O2–) from molecular oxygen, which then decomposes to hydrogen peroxide (H2O2) and singlet oxygen. In turn, H2O2 can react with (O2–) to form hydroxyl radicals. The potent oxidants contained within the phagosome can denature bacterial proteins, thereby rendering them susceptible to proteolysis. Additionally, some of the neutrophil proteases are activated by the oxidants. These coordinated events serve to enhance breakdown of pathogens at the site of infection. 
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


