Vitamin B12 and Folic Acid
David Watkins and David S. Rosenblatt
VITAMIN B12
Cobalamin (vitamin B12) is a complex organo-metallic molecule that is synthesized by many bacteria and is obtained in the human diet from meat, fish, and dairy products. It is not present in plant foods, so strict vegetarians are at risk for dietary deficiency. Derivatives of cobalamin are required for the activity of two enzymes: (1) methylcobalamin (MeCbl) is generated during the catalytic cycle of methionine synthase, a cytoplasmic enzyme that catalyzes methylation of homocysteine to methionine (see Chapter 138), and (2) 5′-deoxyadenosylcobalamin (AdoCbl) is required for the mitochondrial enzyme methylmalonyl-CoA mutase to catalyze the conversion of methylmalonyl-CoA, formed during catabolism of branched-chain amino acids and odd-chain fatty acids, to succinyl-CoA (see Chapter 137). Therefore, inborn errors of cobalamin metabolism result in isolated methylmalonic aciduria, isolated homocystinuria, or combined methylmalonic aciduria and homocystinuria, depending on which step in cobalamin metabolism is affected.1-4 In these disorders, hypomethioninemia usually occurs together with hyperhomocysteinemia. Elevated homocysteine levels are associated with an increased risk of thrombosis; decreased methionine is associated with abnormalities of the white matter of the nervous system. Elevated levels of methylmalonic acid are usually associated with metabolic acidosis.
 METABOLISM OF COBALAMIN
METABOLISM OF COBALAMIN 
Absorption of dietary cobalamin is a complex process that is dependent on a cobalamin-binding protein, intrinsic factor (IF), secreted by the parietal cells of the stomach.6 Cobalamin in food exists bound to proteins. After proteins are digested, cobalamin becomes bound in the stomach to haptocorrin, a cobalamin-binding protein secreted in the saliva. Haptocorrin is broken down in the intestine and cobalamin binds the IF. The IF-cobalamin complex is taken up by enterocytes in the distal ileum in a process mediated by a receptor, cubam, composed of the proteins cubilin and amnionless. After uptake by enterocytes, cobalamin is released into the circulation, bound to the transport protein transcobalamin (TC).
TC-bound cobalamin is available for uptake by most cell types. Following endocytosis, mediated by an as-yet uncharacterized cell-surface receptor, the cobalamin-TC complex dissociates in lysosomes, and free cobalamin is transferred to the cytoplasm. Subsequently, cobalamin may become associated with methionine synthase in the cytoplasm, or it may be transported to the mitochondria, where it is converted to adenosylcobalamin and becomes associated with methylmalonyl-CoA mutase.1
 ISOLATED METHYLMALONIC ACIDURIA
ISOLATED METHYLMALONIC ACIDURIA
Metabolic Derangement
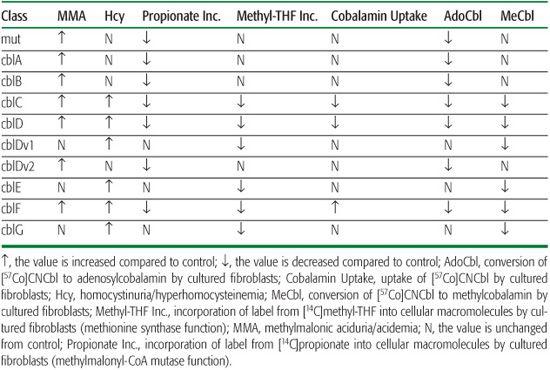
Isolated methylmalonic aciduria is seen in patients with mutations affecting methylmalonyl-CoA mutase itself (the mut disorder [OMIM 251000] due to mutations at the MUT locus on chromosome 6p21; see Chapter 137) or with mutations resulting in decreased synthesis of adenosylcobalamin (the cblA disorder [OMIM 251100], caused by mutations at the MMAA gene on chromosome 4q31.1-31.2, and the cblB disorder [OMIM 251110], caused by mutations in the MMAB gene on chromosome 12q24). The mut disorder has been subdivided into two classes: (1) in mut0 there is no stimulation mutase activity in cultured cells on incubation with hydroxycobalamin (OHCbl), and there is often no detectable mutase protein; (2) in mut−, mutase activity is stimulated by addition of OHCbl.1
Clinical Presentation
Patients with isolated methylmalonic aciduria are prone to episodes of life-threatening acidosis, often in response to infection or to increased protein intake. Initial presentation is most frequently during infancy, with vomiting, hypotonia, irritability, and lethargy, then progressing to coma and death if not treated. In the most severely affected individuals, there may be intractable acidosis during the neonatal period that leads to death, but in many patients the disorder is characterized by recurrent acidotic crises characterized by severe ketoacidosis, hyperammonemia and hyperglycemia, or hyperglycemia with anemia, neutropenia, thrombocytopenia or pancytopenia, failure to thrive, or metabolic stroke. Prolonged elevation of plasma methylmalonic acid may lead to end-stage renal failure necessitating transplantation. Studies of series of patients have shown that presentation is typically most severe in individuals with mut0, while cblB is less severe and cblA and mut− patients have the least severe presentation and the best prognosis.13,14
Diagnostic Tests
Organic acid analysis in these patients reveals elevated levels of methylmalonic acid and of metabolites such as methylcitric acid, 3-hydroxypropionic acid, and tiglylglycine. Tandem mass spectroscopy of acylcarnitines shows elevated propionyl carnitine. Elevated glycine may be present in blood and urine. Elevation of methylmalonic acid levels may also be seen in individuals with methylmalonyl-CoA epimerase (racemase) deficiency and in patients with mutations affecting either subunit of succinyl-CoA ligase; however, in these orders, the levels are lower than in mut, cblA, and cblB. 
Genetics
All three disorders are inherited as autosomal recessive traits.
 ISOLATED HOMOCYSTINURIA
ISOLATED HOMOCYSTINURIA
Metabolic Derangement
Homocystinuria in the absence of methylmalonic aciduria is seen in patients with the cblE (OMIM 250940) and cblG (OMIM 236270) disorders. In both disorders, methionine synthase function is impaired; in the cblG disorder, synthase-specific activity in cell extracts is decreased under all assay conditions, while in the cblE disorder, specific activity is decreased only in the presence of limiting concentrations of exogenous reducing agent.19 The cblG disorder is caused by mutations affecting the MTR gene on chromosome 1q43, which encodes methionine synthase.20,21 The cblE disorder is caused by mutations of the MTRR gene on chromosome 5p15.3-15.2, which encodes methionine synthase reductase.22
Clinical Presentation
Patients with these disorders are characterized by megaloblastic anemia and neurological problems, including developmental delay, cerebral atrophy, hypotonia, microcephaly, and seizures. Homocystinuria is less severe in patients with these disorders than in patients with homocystinuria due to cystathionine β-synthase deficiency. Presentation is typically within the first 2 years of life but can present in adulthood as well.23
Diagnostic Tests
Decreased methionine synthase activity results in increased serum total homocysteine and in homocystine in the urine, combined with reduced levels of methionine and elevated cystathionine. Serum cobalamin levels are normal. These disorders can be differentiated from homocystinuria due to cystathionine β-synthase deficiency, which presents with higher levels of homocysteine, elevated methionine, and decreased cystathionine levels (see Chapter 138, Table 138-1). Fibroblasts are characterized by decreased methyl-THF incorporation with normal propionate incorporation and by decreased methylcobalamin synthesis with normal adenosylcobalamin synthesis. The disorders can be differentiated by the response of methionine synthase–specific activity to the titration of a reducing agent but more typically are differentiated by complementation analysis.
Genetics
Both disorders are inherited as autosomal recessive traits. 
 COMBINED METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA
COMBINED METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA
Metabolic Derangement
Three inborn errors of metabolism that affect early steps in cobalamin metabolism result in combined methylmalonic aciduria and homocystinuria. The cblC disorder (OMIM 277400) is caused by mutations at the MMACHC locus on chromosome 1p34.1,27 and the cblD disorder (OMIM 277410) is caused by mutations at the MMADHC locus on chromosome 2q23.2.28 The function of neither of these gene products is understood. Although the original patients with the cblD disorder had combined methylmalonic acid and homocystinuria, additional patients with isolated homocystinuria (cblD variant 1) and isolated methylmalonic aciduria (cblD variant 2) have subsequently been recognized.29 The cblF disorder results in inability to transfer endocytosed cobalamin from the lysosome to the cytoplasm.30 The gene underlying this disorder has not yet been identified.
Clinical Presentation
The large majority of patients with combined methylmalonic aciduria and homocystinuria belong to the cblC class, with over 300 patients identified. Individuals with this disorder typically present during the first year of life with megaloblastic anemia, failure to thrive, developmental delay, hypotonia, seizures, macrocephaly, and cerebral atrophy. Despite the presence of methylmalonic aciduria, metabolic decompensation is infrequent. Other patients present later in life or in adulthood with ataxia, dementia, or psychosis. A distinctive salt-and-pepper retinopathy has been described in some cblC patients. Presentation in classic cblD disease is similar to that of cblC. Presentation of the cblF disease is variable. Frequent findings have included feeding difficulties, failure to thrive, growth retardation, and persistent stomatitis.
Diagnostic Tests
Patients with these disorders have the biochemical characteristics of both methylmalonic aciduria and homocystinuria, although methylmalonic acid levels are generally lower than in patients with isolated methylmalonic aciduria. Fibroblasts show decreased function of both methylmalonyl-CoA mutase and methionine synthase, and decreased synthesis of both adenosylcobalamin and methylcobalamin (Table 147-1). Fibroblasts from patients with the cblC or cblD disorder are unable to accumulate cobalamin in cells, reflecting the inability of cells to retain cobalamin that is not bound to one of the cobalamin-dependent enzymes. In cblF disorder, fibroblasts accumulate large amounts of cobalamin, but most of this is unmetabolized cobalamin that is trapped within the lysosomes.
Genetics
Both disorders are inherited as recessive autosomal traits. 
Treatment
Patients with inborn errors of cobalamin are treated with large doses (up to 1 mg per day) of cobalamin administered intramuscularly. In the cblC disorder, hydroxycobalamin is more effective than CNCbl, the standard form of pharmacological vitamin B12)32; this may be true for other disorders as well. In patients with methylmalonic aciduria, protein restriction represents an important aspect of treatment. Supplementation with carnitine and with antibiotics (to reduce propionate production by gut bacteria) has also been used. Supplementation with betaine, which supports conversion of homocysteine to methionine by the liver enzyme betaine-homo-cysteine methyltransferase, has proved effective in patients with homocystinuria.
 INBORN ERRORS OF COBALAMIN UPTAKE AND TRANSPORT
INBORN ERRORS OF COBALAMIN UPTAKE AND TRANSPORT
Combined methylmalonic aciduria and homocystinuria is also seen in patients with defects affecting intestinal uptake of cobalamin and its transport in the serum. Decreased uptake occurs in individuals with intrinsic factor deficiency (OMIM 261000), caused by mutations at the GIF gene on chromosome 11q13,33,34 and in those with Imerslund-Gräsbeck syndrome (IGS; OMIM 261100). The latter disorder is the result of dysfunction of the cubam receptor in the distal ileum, which can be caused by mutations at the CUBN gene on chromosome 10p12.1 or the AMN gene on chromosome 14q32, which encode the cubilin and amnionless components of cubam; additional IGS families are not linked to either chromosome and represent further genetic heterogeneity in this disorder.35,36 Patients usually present between 1 and 5 years of age with decreased serum cobalamin levels, megaloblastic anemia, and neurological impairment. IGS patients often have proteinuria as well.37 In the absence of readily available clinical tests to assess cobalamin uptake, specific diagnosis in patients with disorders of cobalamin uptake depends on sequencing of the GIF, CUBN, and AMN genes.
In the absence of readily available clinical tests to assess cobalamin uptake, specific diagnosis in patients with disorders of cobalamin uptake depends on sequencing of the GIF, CUBN, and AMN genes.
Table 147-1 Clinical and Laboratory Findings in Inborn Errors of Metabolism
Patients with transcobalamin deficiency (OMIM 275350) usually present in the first months of life with megaloblastic anemia; failure to thrive; weakness; and, frequently, immunologic dysfunction; without treatment, neurological impairment develops.1 Because a majority of cobalamin circulates bound to haptocorrin (which is not available for uptake by most types of cells) rather than to transcobalamin, circulating cobalamin levels may be normal in this disorder.  Treatment involves maintaining very high serum levels of cobalamin by either oral or intramuscular route.
Treatment involves maintaining very high serum levels of cobalamin by either oral or intramuscular route.
FOLIC ACID
The folates are a class of molecules comprising folic acid (pteroylglutamic acid) and its derivatives. Folic acid consists of a pteroic acid (a pteri-dine ring attached to para-aminobenzoic acid) with an attached glutamate residue. Biologically active folates differ from folic acid by reduction of the pterin ring to its tetrahydrofolate (THF) derivative, by the attachment of a variety of one-carbon groups to N5 and N10 of the pterin ring, and by addition of up to six additional glutamate residues by γ-peptide linkage. Folate is synthesized by many types of bacteria and is obtained in the diet from both plant and animal foods.
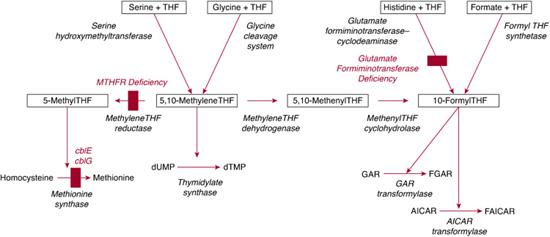
Within cells, folates accept one-carbon units from various sources, primarily serine (via the reaction catalyzed by serine hydroxymethyltransferase) but also from glycine (the glycine cleavage system), formate (formyl-THF synthetase), and histidine (breakdown mediated by glutamate formiminotransferase and formiminotetrahydrofolate cyclodeaminase). Several reactions utilize one-carbon units derived from folates (Fig. 147-1). Methyl-THF is required for methylation of homocysteine to form methionine, catalyzed by methionine synthase (described in the section on vitamin B12; see also Chapter 138). Catalytic conversion of uridylate to thymidylate requires 5,10 methylene THF for the activity of thymidylate synthase. Carbons 2 and 8 of the purine ring are added by the folate-dependent enzymes GAR transformylase and AICAR transformylase, both of which use 10-formyl-THF as a one-carbon donor. Thus, synthesis of both purines and one of two pyrimidines required for DNA synthesis are dependent on folates.3,4,38
 MTHFR DEFICIENCY
MTHFR DEFICIENCY
The most common inborn error of folate metabolism is methylene-THF reductase (MTHFR) deficiency (OMIM 236250) due to mutations of the MTHFR gene on chromosome 1p36.3. Over 90 patients with this disorder have been identified (see also Chapter 138). Decreased MTHFR activity results in deficiency of methyl-THF, the source of the methyl group used by methionine synthase. Thus MTHFR deficiency is characterized by hyperhomocysteinemia and homocystinuria with hypomethioninemia. Clinical presentation varies, but patients usually show symptoms during infancy or early childhood that include feeding difficulties, lethargy, hypotonia, developmental delay, and seizures.39 Cerebral atrophy and demyelination are often present. Patients with later onset may have mental retardation, ataxia, or psychiatric problems. Unlike patients with inborn errors of cobalamin metabolism, in which methionine synthase activity is impaired, patients with MTHFR deficiency do not have megaloblastic anemia. Cultured fibroblasts have decreased incorporation of label from [14C]formate into cellular macromolecules, but incorporation of label from [14C]methyl-THF is normal. Diagnosis depends on enzyme assay of cell extracts demonstrating reduced MTHFR-specific activity. 
Patients with MTHFR deficiency have been treated with a variety of agents, including methyl-THF and other folates, methionine, pyridoxine, cobalamin, carnitine, betaine, and riboflavin, individually and in various combinations. Of these, betaine appears to be the most effective. Treatment is most successful when the disease is diagnosed at an early stage, before irreversible neurological damage can occur. 
 GLUTAMATE FORMIMINOTRANSFERASE DEFICIENCY
GLUTAMATE FORMIMINOTRANSFERASE DEFICIENCY
A small number of patients have been identified who have deficiency of the bifunctional enzyme that catalyzes the transfer of the formimino group from formiminoglutamate (FIGlU), generated during histidine catabolism, to tetrahydrofolate (THF) to form formimino-THF, which is then converted to 10-formyl-THF. Patients with glutamate formiminotransferase deficiency (OMIM 229100) are characterized by elevated FIGlu levels either constitutively or in response to a histidine load. Because the affected enzyme is expressed only in liver and kidney, using enzyme assay to confirm the diagnosis is rarely possible. Two classes of patients have been reported: one was characterized by mental retardation, physical retardation, and cortical atrophy, while the second showed no mental retardation but massive FIGlu excretion. However, it has been suggested that the severe manifestations in the first group of patients were the result of ascertainment bias.43
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


