Vascular and Lymphatic Anomalies
Vascular anomalies are broadly divided into two groups based on biologic and clinical behavior: vascular tumors and vascular malformations.1 Vascular tumors are true neoplasms that arise from cellular hyperplasia. In contrast, vascular malformations are congenital lesions originating from errors of embryonic development and exhibit normal endothelial cell turnover.1 Historically, the field of vascular anomalies has been hindered by a myriad of confusing and misused terminology and nomenclature. This, along with the rarity and often complex nature of some of these disorders, has combined to make diagnosis and treatment of vascular anomalies difficult. However, the last several decades have brought better insight and understanding into the field of vascular anomalies, with improved knowledge of blood vessel angiogenesis and the development of a more logical classification system.
Classification
In 1996, the International Society of the Study of Vascular Anomalies formally accepted the biological classification system in use today (Table 72-1).2,3 This system divides these anomalies into vascular tumors and vascular malformations based on physical characteristics, natural history, and cellular features. Examples of vascular tumors are infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). Vascular malformations can be divided based on vascular channel type (capillary, lymphatic, venous, arterial, or combined) or by flow (slow or fast). Examples of slow-flow lesions are capillary malformations (CM), lymphatic malformations (LM), and venous malformations (VM). Fast-flow lesions include arteriovenous fistulas (AVF) and arteriovenous malformations (AVM).

Nomenclature
In the 19th century, Virchow first described the histologic features of vascular nevi.4 He initiated the term ‘angioma’, which became the default term to describe all such nevi regardless of natural history or other clinical features. He also labeled the IH ‘angioma simplex’, a lesion that has been historically referred to as ‘capillary hemangioma’ and ‘strawberry hemangioma.’ Virchow’s ‘angioma cavernosum’ was used to label two distinct lesions, IH (when located deep to the skin) and VM, because both have similar appearance on physical examination. ‘Angioma racemosum’ was Virchow’s designation for what today is termed an arteriovenous malformation and which has previously been called an ‘arteriovenous hemangioma.’
Wegener, a student of Virchow, described the histology of LMs, which he called ‘lymphangiomas.’5 The classic term ‘cystic hygroma’, referring to LM, unfortunately also continues to have common usage. Thus, both the terms cystic hygroma and lymphangioma should be abandoned in favor of LM (macrocystic and microcystic, respectively). The problems with this jumble of descriptive and histologic terms are obvious. The same lesion can often have several different names, and simultaneously, the same name can refer to several different lesions. For example, the term ‘hemangioma’, combined with descriptive modifiers such as ‘strawberry’, ‘cavernous’, and ‘lympho-’, is used to describe tumors, birthmarks, and vascular malformations alike. Vascular anomalies with quite distinct features, whether congenital or acquired, or whether they spontaneously regress or progress over time, become lumped under the umbrella term ‘hemangioma.’ These faulty designations lead to improper diagnosis and treatment for patients as well as leading to misguided interdisciplinary communication and research efforts.
Vascular Tumors
Infantile Hemangioma
IH are the most common tumor of infancy. They occur in about 4% of infants, though early studies were as high as 10%, probably due to the inclusion of other vascular lesions.6 The incidence is higher in premature infants, Caucasians, and females (by a 3 to 5 : 1 ratio). 6,7 Advanced maternal age, multiple gestations, and placental abnormalities are also risk factors.8 IH have a unique and characteristic life cycle consisting of three phases: proliferative, involuting, and involuted.
Pathophysiology
The precise etiology of IH remains unknown. Viral causes have been speculated, but none elucidated. Some studies suggest that they arise from the clonal expansion of endothelial stem/progenitor cells, the source of which is unclear.9–11 One report concluded these cells arise from a population of resident angioblasts, arrested in an early stage of vascular development.12 As hemangiomas are more common in females, estrogen, which has a stimulatory effect on endothelial cells, may factor in the development of these lesions. Its receptors are present on endothelial cells and elevated levels of estradiol have been found in infants with hemangiomas.13,14
The expression of placental markers (including CD 32, Fcγ-RIIb, glucose transporter 1 [GLUT-1], indoleamine deoxygenase [IDO], insulin growth factor 2 [IGF-2] Lewis Y antigen, merosin, type II 17-hydroxysteroid dehydrogenase [17HSDβ2], tissue factor pathway inhibitor 2 [TFPI-2], and type III iodothyronine deoidinase) by hemangioma endothelial cells suggests a placental origin.15–21 GLUT-1, an erythrocyte type glucose transporter, is a specific marker for endothelial cells of hemangiomas, but is not found in other vascular anomalies.15 The placental cells may arrive at fetal tissue following local placental disruption as an embolic nidus though the permissive right to left shunt of fetal circulation. This may occur during chorionic villus sampling or placental complications such as pre-eclampsia and placenta previa, which have shown to be predisposing factors for hemangiomas.8,22,23
The dysregulation of angiogenesis can be seen during the proliferation and involution of hemangiomas, and is suspected to be a cause of the disease. IH in the proliferative phase express high levels of fibroblast growth factor (FGF), TIE-2, angiopoietins, matrix metalloproteinases (MMPs) and vascular endothelial growth factor A (VEGF-A) and its receptor (VEGFR), all of which play critical roles in the formation of blood vessels during and after embryogenesis.24–30 The tumor during this phase is composed of plump, rapidly dividing endothelial cells forming a mass of sinusoidal vascular channels. Enlarged feeding arteries and draining veins often vascularize the tumor. Markers for mature endothelium, CD-31 and von Willebrand factor, are present on these neoplastic endothelial cells. Involuted hemangiomas express normal levels of these factors, but elevated levels of tissue inhibitor of TIMP1, a metalloproteinase that inhibits new blood vessel formation, and interferon-β.28,29 The endothelial cells of the tumor flatten as apoptosis progresses, the vascular channels dilate, and the tumor assumes a lobular architecture with replacement by fibrofatty stroma.31 All that remains in the involuted phase is a residuum of fibrofatty tissue with tiny capillaries and mildly dilated draining vessels.
Clinical Features
IH are not fully developed at birth and first appear in the neonatal period with a median age of onset of 2 weeks. A premonitory cutaneous sign may be present at birth in 30–50% of cases.1 They are most often cutaneous (80%) and have an anatomic predilection for the head and neck region (60%). They occur in the trunk and extremities 25% and 15% of the time, respectively.2 Internal and visceral lesions are uncommon. Up to 20% of patients can have multiple lesions, and these cases are most likely to have internal involvement affecting organs such as the liver and gastrointestinal (GI) tract.1
The proliferative phase of IH is marked by rapid growth for the first six to eight months that typically plateaus by age 10–12 months. Tumors that involve the superficial dermis present as a red, raised lesion (Fig. 72-1A). Superficial tumors that are larger or that exhibit more rapid growth can occasionally cause ulceration of the skin with bleeding. Tumors in the lower dermis, subcutaneous tissue, or muscle appear bluish in color with slightly raised overlying skin (previously incorrectly termed ‘cavernous’ hemangiomas). With experience, history and physical examination can establish an accurate diagnosis for most of these tumors. The involuting phase of hemangiomas occurs from age 1 to 7 years during which time the tumor slowly regresses, although it may grow in proportion with the child. This phase is notable for fading color of the tumor from crimson to a dull purple, accompanied by a deflation of the tumor mass (Fig. 72-1B).1 The skin may become pale, usually in the center of the tumor first, spreading outwards. Fifty per cent of tumors have completed involution by 5 years of age, and 70% by age 7 years. There is continued gradual improvement in these aspects until the regression is entirely complete by age 10–12 years.32 In the final involuted phase of the tumor, 50% of patients have nearly normal skin in the area of the prior lesion. Patients with larger tumors can have lax or redundant skin and yellowish discoloration. Scars will persist if parts of the tumor were previously ulcerated.1
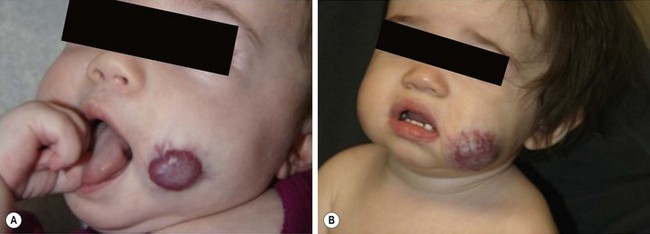
FIGURE 72-1 (A) This infant has an infantile hemangioma that is in the proliferative phase. This hemangioma was not present at birth but was noted at several weeks of age. (B) This hemangioma is in its involuting phase.
The differential diagnosis of cutaneous hemangiomas consists primarily of other vascular anomalies. CM that involves the skin can be mistaken for superficial hemangiomas, or vice versa. Deeper hemangiomas can be confused for VM or LM as all can appear as bluish masses through the skin. Hemangiomas with fast-flow vascularity of the parenchyma could be confused for AVM, but the age of onset and history generally distinguishes the two. Congenital hemangiomas, discussed in a later section, may be misdiagnosed as vascular malformations, which are congenital by definition. Pyogenic granulomas may be differentiated from hemangiomas by their rare appearance before six months (mean age is 6 to 7 years), and their frequent association with minor trauma.33,34 Other tumors such as TA, hemangiopericytoma, and fibrosarcoma can also be confused.35
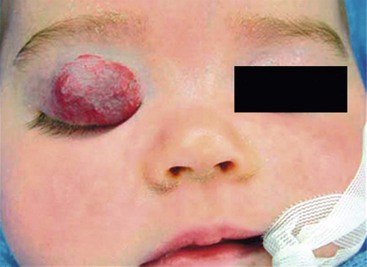
The primary local complications that can occur with cutaneous hemangiomas are ulceration, bleeding, and pain. Hemangiomas are rarely life-threatening, but complications can be anticipated by recognition of the anatomic distribution of the lesion.36 Lesions in the cervicofacial region can lead to airway obstruction as they grow during the proliferative phase. Very large hemangiomas, notably in the liver, can lead to high-output congestive heart failure (secondary to fast-flow and vascular shunting within the tumor), hypothyroidism, and abdominal compartment syndrome. Facial lesions involving the eyelid, nose, lip, or ear can result in tissue destruction with cosmetic consequences. Periorbital and eyelid hemangiomas can cause visual axis obstruction, leading to deprivation amblyopia (Fig. 72-2). Alternatively, distortion of the cornea can cause astigmatic amblyopia. GI hemangiomas are very rare, but may manifest with GI bleeding.
Associated Congenital Anomalies
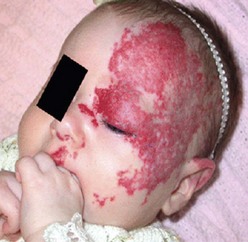
IH are rarely associated with other anomalies. However, a few such anomalies have been described, more commonly with larger and midline hemangiomas. Cervicothoracic hemangiomas can be seen in conjunction with sternal nonunion.37 Tumors of the lumbosacral area have been noted to occur along with spinal dysraphism abnormalities such as meningocele and tethered spinal cord.38,39 Hemangiomas of the pelvis and perineum have been found in association with urogenital and anorectal anomalies. PHACES association describes hemangiomas associated with congenital ocular abnormalities such as microophthalmia, cataracts, optic nerve hypoplasia, posterior fossa cystic malformations, hypoplasia or absence of carotid and vertebral vessels, as well as malformation of the aortic arch (Fig 72-3).40,41 Females are affected in 90% of cases. These patients are at risk for stroke in the neonatal period and migraines in older ages.
Other Manifestations
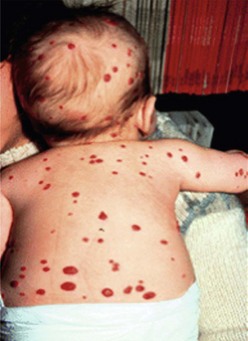
The presence of multiple disseminated hemangiomas is termed hemangiomatosis. The cutaneous tumors are usually tiny (<5 mm) and dome-like. When five or more lesions are present, occult visceral lesions, most commonly in the liver, may also be present (Fig. 72-4). Screening patients with ultrasound (US) and/or magnetic resonance imaging (MRI) may be indicated for these patients.
Imaging
Proper radiologic diagnosis of vascular anomalies is dependent on specific expertise and clinical experience with the radiologic features of these lesions.42 Ultrasound and MRI are the most useful imaging modalities. Ultrasound of proliferative phase hemangiomas demonstrates a mass with dense parenchyma exhibiting fast-flow vascularity.43,44 This distinguishes deep IH from VM, which exhibit slow-flow vascularity and larger blood-filled spaces. MRI of proliferating hemangiomas shows a lobulated solid mass of intermediate intensity with T1 spin-echo sequences, and moderate hyperintensity on T2 spin-echo. Flow voids that represent fast flow and shunting are seen in and around the tumor. During the involuting phase, MRI demonstrates decreased flow voids and vascularity, with the mass taking on a more lobular and fatty appearance.45
Treatment
The majority of IH do not require any specific treatment other than observation and reassurance of the parents.46 Even tumors that exhibit rapid growth or fiery red skin will spontaneously regress and leave behind little to no evidence of their presence. However, regular follow-up is important as the potential complications have few prognostic indicators. Serial photographs are very helpful in documenting progression and subsequent improvement. Reasons for treatment or referral to a vascular anomalies specialist or center include dangerous locations (impinging on a vital structure such as the airway or eye), unusually large size or rapid growth, and local or endangering complications (skin ulceration or high-output heart failure).
Hemangiomas exhibiting the above risk factors or complications should be considered for treatment. Since hemangiomas are tumors of pure angiogenesis, pharmacologic therapy involves angiogenesis inhibition. Systemic corticosteroids, which inhibit the expression of VEGF-A by hemangioma-derived stem cells and thus angiogenesis, were first line therapy for decades.47–49 Oral prednisone is given at a dose of 2–3 mg/kg/day. Doses up to 5 mg/kg/day have been used for life-threatening complications of large hemangiomas causing airway obstruction or heart failure. The overall response rate is 80–90% with initial improvement in the color and tension of the mass usually noted within one week. The steroids are maintained with a very gradual taper every two to four weeks with the goal of discontinuation around age 10–11 months. Live vaccines such as polio, measles, mumps, rubella, and varicella should be withheld while children are taking prednisone. Hemangiomas will have rebound growth if steroids are tapered or stopped too quickly. Return to the initial dosage and slower tapering will usually treat this problem. Potential complications of steroid use in infants and children include impaired growth and weight gain in about one-third of cases. Almost all children will have ‘catch up’ growth and return to pretreatment growth curves by age 14–24 months.50 Cushingoid facies occur in almost all patients and normalizes upon tapering. In rare circumstances, steroids may induce hypertension or hypertrophic cardiomyopathy, both of which are indications to wean or change therapy.50
Intralesional corticosteroids are used for small cutaneous hemangiomas that cause local deformity or ulceration, especially for lesions of the eyelid, nose, cheek, or lip.51 A total of three to five injections are typically given at intervals of six to eight weeks at a dose of 3–5 mg/kg/injection.52 The response rate approaches that of systemic steroids. Subcutaneous atrophy is a potential complication of steroid injection, but is usually temporary. There have been reported cases of blindness following intralesional steroid injection for periorbital hemangiomas.53 This is presumed to be secondary to particle embolization into the retinal artery through feeding vessels. Manual compression around the periphery of the tumor is recommended during injection to minimize embolization through draining veins.
Propranolol, a nonselective beta blocker, has recently been recognized as an important treatment option for hemangiomas and in most centers has become first line pharmacotherapy. A child with a nasal capillary hemangioma treated with propranolol for steroid-induced cardiomyopathy had regression of his lesion.54 This revelation led to the publication of several more studies supporting this finding.55–58 Propranolol is given orally at 2–3 mg/kg/day, in two or three divided doses, and discontinued following regression of the lesion.55,59 Treatment often leads to a consistent, rapid, therapeutic effect with softening of the lesion on palpation and color shift from intense red to purple.55 Propranolol is well tolerated but can cause rare side effects such as bradycardia, gastroesophageal reflux, hypoglycemia, hypotension, rash, somnolence, and wheezing.55,56,58–61 Several mechanisms of action have been proposed.62 Propranolol inhibits β-adrenoreceptors, which are activated by adrenaline, causing vasoconstriction of capillaries supplying the hemangioma.63,64 This likely leads to the visible changes in color and palpable softening. Blockage of β-adrenoreceptors also results in decreased expression of VEGF and MMPs, thereby inhibiting angiogenesis, and induces apoptosis in endothelial cells.65,66
Recombinant interferon was once considered as a second line agent, but has fallen out of favor except in very limited circumstances.67–71 A small subset of patients (5–12%) may develop a severe complication known as spastic diplegia.72,73 Spasticity usually resolves if the drug is terminated quickly. Children receiving IFN should be followed carefully by a neurologist. Though experience is limited, low-dose, high frequency anti-angiogenic regimens using vincristine can be effective.74 The use of interferon and vincristine has waned since the introduction of propranolol therapy.
Although attractive in concept, laser therapy is not often beneficial for IHs, except for a few specific indications.75 The flash lamp pulse-dye laser penetrates the dermis to a depth of only 0.75–1.2 mm. Most cutaneous hemangiomas are deeper than this, and therefore not affected by laser treatment. Additionally, laser therapy carries risks of scarring, skin hypopigmentation, and ulceration, which may lead to a poor result compared to observation alone. One instance in which the laser is advantageous is the treatment of telangiectasias that often remain in the involuted phase of hemangioma. The use of endoscopic continuous wave carbon dioxide laser has been shown to be a good strategy for controlling proliferative phase hemangiomas in the unilateral subglottic location.76 Lastly, intralesional photocoagulation with bare fiber Nd:YAG laser can be useful for hemangiomas in certain locations, such as the upper eyelid when visual obstruction is a concern.
Indications for resection of IH vary with patient age. During the proliferative phase in infancy, well-localized or pedunculated tumors can be expeditiously resected with linear closure, especially for tumors complicated by bleeding and ulceration. Sites that are most amenable to resection are the scalp, trunk, and extremities. Other modalities to treat ulceration include wound care with dressing changes, topical antibiotics, and topical steroids, which can accelerate healing.77 Tumors of the upper eyelid that obstruct vision and that do not respond to pharmacologic therapy may also require excision or debulking. Focal lesions of the GI tract with bleeding that fail medical management may require enterotomy and resection, or endoscopic band ligation. Diffuse, patchy involvement is the more common presentation of GI hemangiomas. Management is difficult but most lesions eventually involute and stop bleeding.78 Preoperative localization with capsule endoscopy and/or intraoperative endoscopy may be necessary to identify lesions in the small bowel.79
During the involuting phase, resection may be needed for hemangiomas that are large and protuberant, and therefore likely to create excess and lax overlying skin.80 Indications for resection include: (1) it is obvious that resection will be necessary sooner or later; (2) the surgical scar will be identical regardless of timing of operation; and (3) the surgical scar can easily be hidden. Lesions of the nose, eyelids, lips, and ears require special expertise. It is often preferable to perform the operation for the above indications during the preschool years before children become aware of and focus on body image differences that may lead to low self-esteem.
Finally, for the difficult to treat and life-threatening large hemangiomas, especially in the liver, angiographic embolization may be required to manage high-output cardiac failure. Arterial catheterization in infants carries significant risks and should generally be limited to those situations with cardiac compromise in which there is the capacity and intent to perform simultaneous embolization. In these rare cases, anti-angiogenic pharmacotherapy remains the first line of therapy and should continue along with angiographic procedures. Repeat embolization procedures may be required. Success with embolization is dependent on occlusion of macrovascular shunts within the tumor rather than occlusion of feeding vessels.81
Congenital Hemangioma
Two types of rare congenital hemangiomas exist that are fully developed at birth and that do not usually exhibit postnatal tumor growth.82 These have been termed rapidly involuting congenital hemangioma (RICH) and the noninvoluting congenital hemangioma (NICH) (Fig. 72-5). Lesions are solitary and affect both genders equally. Unlike IHs, they do not stain positive for GLUT-1.83 The diagnosis is typically made on physical exam at birth, although some can be diagnosed prenatally as early as 12 weeks of gestation.82 Most do not require therapy.
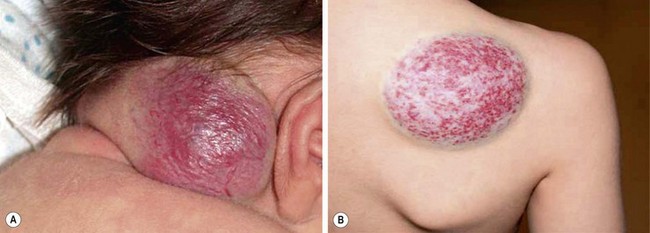
FIGURE 72-5 These two patients are examples of rapid involuting congenital hemangiomas (RICH) and noninvoluting congenital hemangiomas (NICH). (A) The newborn baby has a RICH. This lesion was present at birth and will spontaneously regress, much more quickly than the typical infantile hemangioma. (B) This 9-year-old child has a NICH. This hemangioma has not resolved. If treatment is needed, arterial embolization may be beneficial as these lesions have significant flow to them.
As opposed to IH, RICH is more common on the extremities. Also, RICH will spontaneously regress, much more quickly than IH, by 6 to 14 months; NICH do not involute and grow with the child.82 RICH appear raised and violaceous, often with a central depression or scar, ulceration, telangiectasia, or surrounding pale rim.82,83 MRI will show enhancing hyperintense masses, high-flow vessels within and adjacent to the mass, and the presence of vascular flow voids on T2-weighted imaging.83 NICH are well-circumscribed, plaque-like lesions, often with coarse telangiectasias, areas of pallor, or a white to bluish rim.84 They appear on MRI as homogeneous lesions with isointense signal on T1-weighted imaging and hyperintense on T2-weighted sequences. Both lesions are fast flow on Doppler ultrasound. If treatment is needed for NICH, arterial embolization may be beneficial as these lesions have significant flow to them. Operative excision is reserved for cases with equivocal diagnosis, poor cosmetic appearance, or for complications such as ulceration, bleeding, obstruction, or congestive heart failure.83,85
Hepatic Hemangioma
Hepatic hemangiomas (HH) in infants should be differentiated from ‘hepatic hemangiomas’ that present in adulthood.85 Adult ‘hepatic hemangiomas’, which are sometimes called ‘cavernous hemangiomas’, are in fact VMs. In contrast, HH of infancy are true tumors and have a similar pattern of involution to cutaneous IH. Contrary to popular belief, not all liver hemangiomas are life-threatening. The classic triad of heart failure, anemia, and hepatomegaly is unusual, and most involute without long-term sequelae.
The majority of HH can be divided into three categories: focal, multifocal, and diffuse, each with distinct features (Fig. 72-6).86 Focal HH are the hepatic equivalent of the cutaneous RICH, and also do not stain positive for GLUT-1. They are histologically distinct from the typical IH.15,87 They are fully grown at birth and regress much faster than IH. Many focal lesions are detected antenatally on prenatal ultrasound, or are discovered as an abdominal mass in otherwise healthy infants.88 They are usually asymptomatic, found in equal numbers in both genders, and rarely associated with cutaneous hemangiomas. Transient anemia and moderate thrombocytopenia, due to intralesional thrombosis, are observed in some infants and generally resolve spontaneously. This is in contradistinction to the profound thrombocytopenia seen with Kasabach–Merritt phenomenon (KMP). However, a subset of focal hepatic RICH-type hemangiomas will have macrovascular shunts from the hepatic arteries and/or portal veins to the hepatic veins. These shunts can cause a large steal, accounting for blood-flow demands above and beyond the hypervascular tumor parenchyma, and can result in high-output cardiac failure. These shunts may close as the tumors involute. However, cardiac strain can often mandate interruption of the shunts via embolization. Steroids have been used for solitary focal hepatic lesions with in-utero cardiac failure/cardiomegaly.88,89 However, these lesions may have undergone rapid spontaneous involution and the benefit of steroids remains unproven, though still should be attempted before employing more invasive therapies. Resection is rarely indicated.
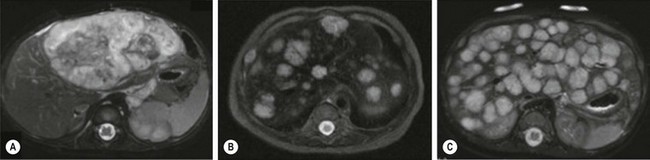
FIGURE 72-6 Most hepatic hemangiomas can be divided into three categories: focal, multifocal, and diffuse. (A) A large focal hepatic hemangioma. (B) The scan depicts multifocal hepatic hemangiomas. (C) Diffuse hepatic hemangioma.
Multifocal HH are true IH and biologically distinct from focal HH. They undergo involution similar to cutaneous IH and stain positive for GLUT-1.87,90,91 They are more common in females and Caucasians.92 Infants with multifocal lesions are typically born earlier and are asymptomatic, but are diagnosed later than focal lesions. These patients often have cutaneous IH and are identified following screening for visceral hemangiomas. Some of these patients will have hypothyroidism. Thus, a serum TSH should be checked in patients with multifocal disease. Infants who are asymptomatic should be observed; however, some can have high-output cardiac failure from macrovascular shunting. Treatment with steroids or propranolol can often close these shunts.54,55 Embolization can be employed in those who fail medical therapy. Serial abdominal ultrasound should be performed in infants with multifocal HH until the lesions involute.
Diffuse HH are also true IH, but are far more serious lesions with a more difficult clinical course. Like multifocal lesions, diagnosis occurs in the weeks following birth. They are more common in Caucasians and females, are associated with cutaneous IH and follow a similar course of involution, and express GLUT-1.92 These patients all develop severe hypothyroidism from high levels of type 3 iodothyronine deiodinase, which inactivates circulating thyroid hormones.20 Aggressive exogenous thyroid hormone replacement is essential to prevent hypothyroid complications such as mental retardation and cardiac failure. Involution of the lesions will usually result in amelioration of the hypothyroidism.93 These innumerable lesions often almost completely replace the normal hepatic parenchyma. Those with considerable disease may have respiratory compromise from compression of the diaphragm and thoracic cavity, but can also develop abdominal compartment syndrome and multisystem organ failure. Aggressive pharmacotherapy with propranolol, steroids, and occasionally low-dose vincristine is warranted in those with cardiac failure, hemodynamically significant shunting, or hypothyroidism to hasten involution.55,86,94–96 Hepatic transplantation is the last resort for critically ill infants.86
Tufted Angioma and Kaposiform Hemangioendothelioma
These vascular tumors of childhood are more aggressive and invasive than IH. TA and KHE probably exist within the same spectrum as they share many overlapping clinical and histologic features.21,97 Both tumors typically present at birth, although they occur postnatally as well. Males and females are affected equally. The tumors are unifocal and are most often located on the trunk, shoulder, thigh, or retroperitoneum. TA present as erythematous macules or plaques and histology reveals small tufts of capillaries.97 KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying and surrounding ecchymosis (Fig. 72-7). The natural history of these tumors is one of continued proliferation into early childhood followed by subsequent, but incomplete regression. These tumors usually persist, albeit in a smaller form.98 Fortunately, they are usually asymptomatic in later stages, though may cause musculoskeletal pain. Histology of these lesions reveals infiltrating sheets of spindle-shaped endothelial cells in the form of irregular lobules, sheets, and lacy network.99 Imaging of KHE depicts an enhancing lesion on MRI with poorly defined margins that extend across tissue planes. This is in contrast to IH, which are well-circumscribed and respective of tissue planes.
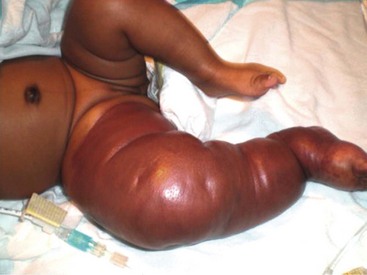
FIGURE 72-7 This infant has a kaposiform hemangioendothelioma (KHE). KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying ecchymosis. These lesions usually regress later in childhood, although usually not completely.
Kasabach–Merritt Phenomenon
KMP was first reported in 1940 in a case of profound thrombocytopenia, petechiae, and bleeding in conjunction with a ‘giant hemangioma.’100 As with many terms in the field of vascular anomalies, this term has been often misused in connection with coagulopathy and other vascular lesions, most prominently VM. However, the profound and persistent thrombocytopenia that occurs with KMP does not occur with either VM or IH. The only known true associations are with TA and KHE.99,101,102 The platelet count with KMP is typically under 10,000/μL, and may be associated with decreased fibrinogen levels, increased D-dimer, and mildly elevated partial prothrombin time (PT) and partial thromboplastin time (PTT). Bleeding can result from this platelet trapping coagulopathy at many sites, including intracranial, GI, peritoneal, pleural, and pulmonary. A microangiopathic hemolytic anemia is also present. Treatment for KHE with KMP is primarily medical as the tumor is usually too large and extensive to be resected. Corticosteroids and interferon-alfa have been effective in about 50% of cases; actinomycin, anti-platelet therapy, cyclophosphamide, doxorubicin, gemcitabine, propranolol, sirolimus, and vincristine have also been found to be beneficial in several case series, as single drugs or in combination, but none of these agents have been shown to be consistently successful.103–111 Platelet transfusions are ineffective and should be avoided unless there is active bleeding. Additionally, heparin may stimulate tumor growth and worsens the thrombocytopenia of KMP and should likewise be avoided. Mortality rates with KHE and TA remain high at 20–30%. KHE not associated with KMP can be followed without treatment as long as the size and location of tumor are limited.
Vascular Malformations
Vascular malformations are congenital lesions of vascular dysmorphogenesis that can be local or diffuse. The majority of vascular malformations are sporadic, though some rare varieties are familial.112–114 They occur in 1.5% of the population.115 Vascular malformations probably result from genetic mutations that lead to dysfunction in the regulation of endothelial proliferation and apoptosis, cellular differentiation, maturation, and cell-to-cell adhesions.116
Embryology and Development of the Vascular and Lymphatic Systems
The vascular system develops during embryogenesis through the processes of vasculogenesis, the formation of new vascular channels from mesodermally derived endothelial precursor cells (hemangioblasts), and angiogenesis, the formation of new blood vessels from preexisting blood vessels. The destiny of endothelial precursors to create different types of blood vessels appears to be imprinted early in embryogenesis by unique cell surface markers.117 The differentiation of angioblasts into an early vascular plexus leads to the creation of primitive blood vessels.118 Following formation of the primary vascular plexus, endothelial cells proliferate and sprout or split from their vessel of origin to form new capillaries. A process called ‘pruning’ then occurs, during which the vascular plexus is remodeled into a system with larger and smaller vessels. The signals for microvascular endothelial cells to proliferate and differentiate for vessel development are controlled by a number of growth factors and their receptors, including VEGF, basic fibroblast growth factor 2 (FGF-2), and angiopoietin 1 (Ang-1).119
The lymphatic system develops around midgestation after the blood vasculature forms, and is thought to derive from either venous endothelial cells or mesenchymal progenitor cells.120–123 It is a one-way valve system that collects fluid and macromolecules from tissue. In 1902, Sabin described the prevailing model of lymphangiogenesis in which venous endothelial cells commit to become lymphatic endothelium and then migrate and proliferate to form lymph sacs.122 These sacs then form a lymphatic plexus, which remodel and mature into the lymphatic vasculature. 120,123 Venous endothelial cells along the anterior cardinal vein capable of differentiating into lymphatic endothelial cells (LECs) begin to express lymphatic endothelial hyaluronan receptor-1 (LYVE-1) on embryonic day (E) 9, a marker specific for lymphatic endothelium.120,123,124 A subgroup of endothelial cells on one side of the vein then begin to express prospero-related homeobox 1 (Prox-1). 125 This transcription factor is required for the maturation and differentiation of LECs.125,126 VEGFR3, also known as Flt4, plays an important role in lymphatic development as well. VEGFR is expressed in both blood and lymphatic vasculature in early embryogenesis, but is restricted to mostly lymphatic vessels later in development127–129 VEGFR3 knockout mice die on E9 with major venous anomalies before any lymphatic sprouting has occurred.116 In contrast, transgenic mice that overexpress the ligand for VEGFR3 (VEGF-C) will develop distended lymphatic channels.130
Capillary Malformations
CM, previously referred to as ‘port-wine stains’, are present at birth as permanent flat, pink-red cutaneous lesions (Fig. 72-8). In the newborn nursery, CM can be confused with nevus flammeus neonatorum, commonly called ‘angel’s kiss’ when located on the face, or ‘stork bite’ when in the posterior cervical location. However, these discolorations are due to transient dilations of dermal vessels and fade with time, whereas CM does not. CM occur with equal gender distribution in 0.3% of infants.131 Multiple CM are rare. The majority of CM appear sporadically, but some familial forms exist that are inherited in an autosomally dominant fashion.132 Capillary malformation–arteriovenous malformations (CM-AVM) are associated with mutations in RASA1, a gene coding for p120-RasGTPase.133 Histologically, cutaneous CM consist of dilated capillary- to venule-sized vessels located in the superficial dermis, with a paucity of surrounding normal nerve fibers.134 These abnormal vessels gradually dilate over time leading to darkening color and occasionally nodular ectasias. CM can also be found in complex-combined vascular malformations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


