Biliary Atresia
Biliary atresia is a relatively rare obstructive condition of the bile ducts causing neonatal jaundice. There is a variable incidence around the world (eg, Europe, 1 in 18,000 live births; France, 1 in 19,500 live births; the UK and Ireland, 1 in 16,700 live births; Sweden, 1 in 14,000 live births; and Japan, 1 in 9640 live births),1–6 with the highest recorded incidence in French Polynesia (1 in 3124 live births).7 There is a slight female predominance.
Biliary atresia first appeared as a distinct entity in the Edinburgh Medical Journal in 1891.8 The concept of ‘correctable’ and ‘noncorrectable’ forms was introduced in 1916.9 Despite the first successful surgical treatment for the correctable type being reported in 1928, there were only a few long-term survivors over the next three decades, all with the correctable type.10–12
In the 1950s and 60s, a variety of procedures for ‘noncorrectable’ disease were developed, but none provided consistent biliary decompression.13–17 Also, the timing of operative intervention was controversial, with reports of ‘spontaneous’ cures, and second-look explorations for the rather mystical belief that a totally fibrotic extrahepatic ductal system might subsequently become patent.18–22
In 1959, the now common Kasai hepatic portoenterostomy procedure was first described and ended a long, hopeless era for patients with the noncorrectable-type disease.23 Kasai’s original report was in Japanese and received little attention until it was published in English in 1968.24 Although effective bile drainage could be achieved after portoenterostomy in about 50% of patients, early repair was crucial and needed to be performed before the age of 2 months. Conversely, effective bile drainage was observed in only 7% of patients if correction was performed after the age of 4 months.25 Kasai’s portoenterostomy procedure gradually gained popularity in the USA, and by the 1990s, more than 90% of infants with biliary atresia had undergone this procedure.26
Recently, the authors had an opportunity to review an original video of Professor Kasai performing his own portoenterostomy.27 Interestingly, his original portal dissection was actually quite shallow and limited, resulting in a narrow portoenterostomy anastomosis, with sutures placed shallowly at 2 and 10 o’clock where the native right and left bile ducts would have been, probably to minimize microscopic bile duct injury. Although not supported by research, the original technique as performed by Kasai may result in superior outcomes as it focuses specifically on the physiologic and anatomic characteristics of the liver in biliary atresia. The authors currently perform a modified version of Kasai’s original portoenterostomy (KOPE) using the minimally invasive approach.
Pathogenesis
There are syndromic and nonsyndromic forms of biliary atresia.28 Syndromic biliary atresia (also known as the embryonic type) is associated with other congenital anomalies, including interrupted inferior vena cava, preduodenal portal vein, intestinal malrotation, situs inversus, cardiac defects, and polysplenia.29 Syndromic biliary atresia accounts for 10–20% of all cases, and is likely to be due to a developmental insult occurring during differentiation of the hepatic diverticulum from the foregut of the embryo. A possible relation between syndromic biliary atresia and maternal diabetes has been reported.28 Nonsyndromic biliary atresia (also known as the perinatal type) may have its origins later in gestation, and may have a different clinical course, with biliary obstruction being progressive.
Reovirus type 3 infection, rotavirus, cytomegalovirus (CMV), papillomavirus, and Epstein–Barr virus have all been proposed as possible etiologic agents, but conclusive evidence is lacking. In one report, CMV infection was found in four of ten patients with biliary atresia,30 and reovirus infection has been found in the livers of up to 55% of biliary atresia patients versus 10–20% in a control group.31 The identification of viruses in children with biliary atresia is inconsistent in the literature, and several viruses have been used to create animal models that may be valuable for assessing the pathogenesis and treatment of biliary atresia.
Generally, biliary atresia is not considered an inherited disorder. However, genetic mutations that result in defective morphogenesis may be important in syndromic biliary atresia. Transgenic mice with a recessive deletion of the inversin gene have situs inversus and an interrupted extrahepatic biliary tree.32 Mutations of the CFC1 gene, which is involved in left–right axis determination in humans, have been identified in a few patients with syndromic biliary atresia.33 The importance of the macrophage migration inhibitory factor gene, which is a pleiotropic lymphocyte and macrophage cytokine in biliary atresia pathogenesis, has also been reported.34 Other studies have identified abnormalities in laterality genes in a small number of patients with biliary atresia, including the transcription factor ZIC3.35 A high incidence of polymorphic variants in the jagged-1, keratin-8, and keratin-18 genes have also been described in a series of 18 children with biliary atresia.36,37 Taken together, the increased incidence of nonhepatic anomalies in children with biliary atresia and genetic mutations reported in subsets of patients with laterality defects suggest that multiple genes are involved, each affecting a small number of patients.
Intrahepatic bile ducts are derived from primitive hepatocytes that form a sleeve (the ductal plate) around the intrahepatic portal vein branches and associated mesenchyme early in gestation. Remodeling of the ductal plate in fetal life results in the formation of the intrahepatic biliary system. This is supported by similarities in cytokeratin immunostaining between biliary ductules in biliary atresia and normal first-trimester fetal bile ducts.38 These findings suggest that nonsyndromic biliary atresia might be caused by a failure of bile duct remodeling at the hepatic hilum, with persistence of fetal bile ducts poorly supported by mesenchyme.
Several studies have investigated whether bile duct epithelial cells are susceptible to an immune/inflammatory attack because of abnormal expression of human leukocyte antigen (HLA) antigens or intracellular adhesion molecules on their surfaces.39,40 A greater than threefold increase in HLA-B12 antigen has been found in babies with biliary atresia compared with controls, particularly in those with no associated malformations.41 Aberrant expression of class II HLA-DR antigens on biliary epithelial cells and damaged hepatocytes in patients with biliary atresia may render these tissues more susceptible to immune-mediated damage by cytotoxic T-cells or locally released cytokines.42 Increased expression of intercellular adhesion molecule-1 (ICAM-1) has been noted on bile duct epithelium in patients with biliary atresia, a finding that may play a role in immune-mediated damage.40 Strong expression of ICAM-1 also has been found on proliferating bile ductules, endothelial cells, and hepatocytes in biliary atresia. A direct relationship exists between the degree of ductal expression of ICAM-1 and disease severity, suggesting that ICAM-1 might be important in the development of cirrhosis.43
Interest has also focused on co-stimulatory molecules. Two processes are involved in the activation of T lymphocytes by antigen-presenting cells (APC). One relates to the expression of major histocompatibility complex class II molecules, which interact directly with T-cell receptors. The other depends on the expression of B7 antigens on APC, and provides the second (co-stimulatory) signal to T-lymphocytes through CD28.44 In postoperative biliary atresia patients with good liver function, co-stimulatory antigens (B7-1, B7-2, and CD40) are expressed only on bile duct epithelial cells, whereas in patients with failing livers these markers are found on the surfaces of Kupffer cells, dendritic cells, and sinusoidal endothelial cells and in the cytoplasm of hepatocytes.45 This suggests that the biliary epithelium and hepatocytes in biliary atresia are susceptible to immune recognition and destruction. Agents that block or prevent co-stimulatory pathways might offer a new therapeutic approach for reducing liver damage.
Two studies have involved comprehensive molecular and cellular surveys of liver biopsies and found a proinflammatory gene expression signature, with increased activation of interferon-γ, osteopontin, tumor necrosis factor-α, and other inflammatory mediators.46,47 These studies may prove to be helpful in delineating the molecular networks responsible for the proinflammatory response and autoimmunity thought to be involved in the pathogenesis of biliary atresia. However, none of these mechanisms appears to be mutually exclusive. Moreover, it is not clear which signs and symptoms are primary and which are secondary.
In summary, the etiology of nonsyndromic biliary atresia is hypothesized to involve a viral or other toxic insult to the bile duct epithelium that induces the expression of new antigens on the surfaces of biliary epithelial cells.48 Coupled with a genetically predetermined susceptibility that is mediated via histocompatibility antigens, these neoantigens are recognized by circulating T-lymphocytes, resulting in an immune cell mediated, fibrosclerosing response.
Classification
Biliary atresia can be classified by using macroscopic appearance and cholangiography findings into three main categories: atresia of the common bile duct (CBD) (type I), atresia of the common hepatic duct (type IIa) or atresia of the CBD and the common hepatic duct (type IIb), and atresia of all extrahepatic bile ducts up to the porta hepatis (type III) (Fig. 43-1). Most patients have type III. In patients with a patent CBD and cystic duct (correctable type) (5% of cases), the gallbladder can be anastomosed to the porta hepatis, (gallbladder Kasai). In more than 90% of cases, however, no patent extrahepatic ductal structures are found at the porta hepatis (i.e., ‘noncorrectable’ type).49
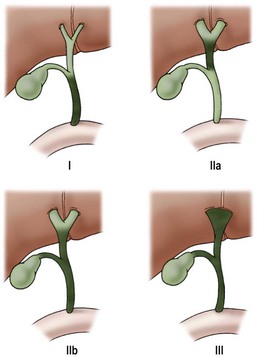
FIGURE 43-1 Morphologic classification of biliary atresia based on macroscopic and cholangiographic findings. Type I, occlusion of common bile duct; type IIa, obliteration of common hepatic duct; type IIb, obliteration of common bile duct, hepatic and cystic ducts, with cystic dilatation of ducts at the porta hepatis, and no gallbladder involvement; type III, obliteration of common, hepatic, and cystic ducts without anastomosable ducts at porta hepatis. (From Lefkowitch JH. Biliary atresia. Mayo Clin Proc 1998;73:90–5.)
Histopathology
Early in the course of biliary atresia, the liver becomes enlarged, firm, and green. The gallbladder may be small and filled with white mucus, or it may be completely atretic (Fig. 43-2). Microscopically, the biliary tracts contain inflammatory and fibrous cells surrounding miniscule ducts, which are probably remnants of the original embryonic duct system. The liver parenchyma is fibrotic and shows signs of cholestasis. Proliferation of biliary neoductules is seen (Fig. 43-3). This process develops into end-stage cirrhosis if adequate biliary drainage cannot be achieved. These early changes are often nonspecific and may be confused with neonatal hepatitis and metabolic diseases.
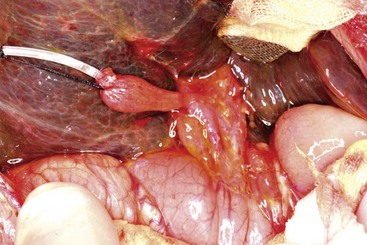
FIGURE 43-2 Type III biliary atresia with an enlarged, firm, green liver and hypoplastic small gallbladder was found in this infant.
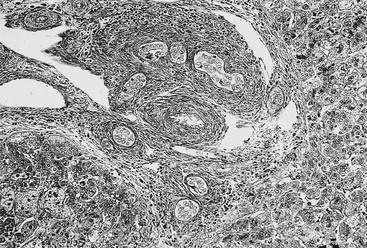
FIGURE 43-3 Photomicrograph of the portal tract of the liver in a 60-day-old infant with biliary atresia. Ductal plate malformation can be seen in the center of the portal space with portal fibrosis. Note ductal metaplasia of hepatocytes (Azan stain, ×100).
It is generally accepted that the pathologic changes seen in biliary atresia are panductal, affecting the intrahepatic biliary tree as well as the extrahepatic bile duct system. Moreover, the intrahepatic bile ducts can be narrowed, distorted in configuration, or irregular in shape.50–52 However, some authors believe that secondary damage occurs only to the extrahepatic biliary system as a result of obliteration of extrahepatic bile ducts during liver formation.53 This theory is strongly supported by the fact that outcome is better if the portoenterostomy is performed early.
The intrahepatic biliary tree is important not only pathologically, but also clinically. The degree of damage that has already occurred in the intrahepatic biliary system is actually responsible for much of the morbidity after hepatic portoenterostomy. Intrahepatic bile duct proliferation likely results from disturbances in formation of the ductal plate as well as ductular metaplasia of hepatocytes.54,55
Certain substances can act as prognostic factors in biliary atresia. Serum levels of interleukin (IL)-6, IL-1ra, insulin-like growth factor-1 (IGF-1), vascular cell adhesion molecule-1 (VCAM-1), and ICAM-1 correlate with liver dysfunction in postoperative biliary atresia patients.43,56,57 Immunohistochemically, a reduction in the expression of CD68 and ICAM-1 at the time of portoenterostomy is associated with a better prognosis.58 The presence of ductal plate malformation in the liver predicts poor bile flow after hepatoportoenterostomy in infants with biliary atresia.59 Growth failure and poor mean weight z-scores three months after hepatoportoenterostomy are also associated with a poor clinical outcome.60
Diagnosis
The cardinal signs and symptoms of biliary atresia are jaundice, clay-colored stools, and hepatomegaly. However, meconium staining is normal in most patients. In the neonatal period, feces are yellow in more than half of patients.61 The newborn’s urine becomes dark brown. Although the neonates are active, and their growth is usually normal for the first few months, anemia, malnutrition, and growth retardation develop gradually because of malabsorption of fat-soluble vitamins. Jaundice that persists beyond two weeks should no longer be considered physiologic, particularly if the elevation in bilirubin is mainly in the direct fraction. Neonatal hepatitis and interlobular biliary hypoplasia are most likely to be confused with biliary atresia. Conventional liver function tests alone are useless for establishing a definitive diagnosis of biliary atresia.
Although a number of diagnostic protocols have been published, the emphasis must always be on early diagnosis (Box 43-1).62,63 The definitive diagnosis of biliary atresia requires further investigations, including special biochemical studies, tests to confirm the patency of the extrahepatic bile ducts, and needle biopsy of the liver. Several authors consider liver biopsy to be the most reliable test for establishing the diagnosis.64,65 Serum lipoprotein-X is positive in all patients with biliary atresia, although it also is positive in 20–40% of patients with neonatal hepatitis. Serum bile acid levels increase in infants with cholestatic disease, but both the total bile acid level and the ratio of chenodeoxycholic acid to cholic acid have no value for differentiating biliary atresia from other cholestatic diseases.66 Hyaluronic acid, which has been considered a serum marker for liver function, has also been reported to be a biochemical marker for evaluating infants with biliary atresia.67 Duodenal aspiration is an easy, noninvasive, and rapid test because biliary atresia can be excluded if bilirubin-stained fluid is aspirated.68 Hepatobiliary scintigraphy with technetium-labeled agents is widely used for differentiating biliary atresia from other cholestatic diseases. In biliary atresia, nucleotide uptake by hepatocytes is rapid, but excretion into the bowel is absent, even on delayed images. In hepatocellular jaundice, isotope uptake is delayed owing to parenchymal disease. Excretion into the intestine may or may not be seen.
Ultrasonography (US) should be performed on all jaundiced infants. Hepatobiliary ultrasound will exclude other surgical causes of jaundice such as choledochal cyst and inspissated bile syndrome. In biliary atresia, the intrahepatic ducts are not dilated on ultrasound because they are affected by the inflammatory process. Various sonographic features have been targeted in an attempt to distinguish biliary atresia from other causes of conjugated hyperbilirubinemia in infants.69–73 In biliary atresia, the gallbladder is small, shrunken, and noncontractile, and there is increased echogenicity of the liver. The presence of other associated anomalies of the polysplenia syndrome is pathognomonic of biliary atresia.74 Differentiation from choledochal cyst and type I biliary atresia also is rapid and simple with ultrasound.75 Irrespective of interobserver variation, failure to visualize the CBD is not diagnostic of biliary atresia because a patent distal CBD can be found in up to 20% of biliary atresia cases. However, an absent gallbladder or one with an irregular outline is suggestive of biliary atresia.73 In some cases, a well-defined triangular area of high reflectivity is seen at the porta hepatis, corresponding to fibrotic ductal remnants (the ‘triangular cord’ sign) (Fig. 43-4).70,71
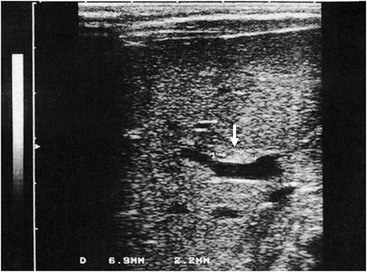
FIGURE 43-4 Ultrasonography shows a well-defined triangular area of high echogenicity (arrow) at the porta hepatis, corresponding to fibrotic ductal remnants (the ‘triangular cord’ sign).
Nonvisualization of the fetal gallbladder may indicate abnormalities ranging from gallbladder agenesis to biliary atresia.76 Amniotic fluid digestive enzymes, which are synthesized by the biliary epithelium, gradually decrease until 24 weeks of gestation. As it is no longer possible to differentiate between abnormally low and physiologically low levels of the enzymes after 24 weeks of gestation, the prenatal diagnosis of biliary atresia is difficult.77
Most patients with biliary atresia can be correctly diagnosed by using an appropriate combination of the investigations just outlined. However, to accurately differentiate biliary atresia, biliary hypoplasia, and severe neonatal hepatitis, cholangiography is usually required.
Recently, laparoscopy-assisted cholangiography has been used to display the anatomic structure of the biliary tree.78 Also, percutaneous cholecystocholangiography may be a useful option to prevent unnecessary laparotomy in infants whose cholestasis is caused by diseases other than biliary atresia.79
Operative Management
Open Surgical Technique
The portoenterostomy procedure for biliary atresia has been repeatedly modified to achieve better rates of jaundice clearance and survival, and hardly resembles Kasai’s original portoenterostomy (Fig. 43-5B). The current favored technique involves an extended lateral dissection around the porta hepatis with a very wide anastomosis (extended portoenterostomy, or EPE) (Fig. 43-5C).80,81
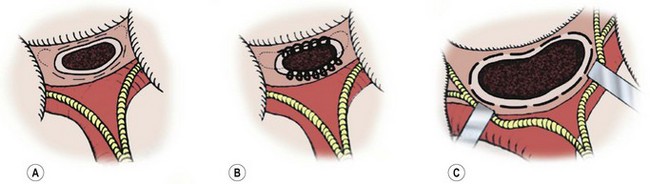
FIGURE 43-5 Salient features of three portoenterostomy procedures are depicted. (A) Modified Kasai portoenterostomy: Interrupted shallow sutures (thin broken lines) are placed in the liver parenchyma around the transected biliary remnant, except at the 2 and 10 o’clock positions, where the right and left bile ducts should be. If sutures are necessary at the 2 and 10 o’clock positions to prevent an anastomotic leak, shallow interrupted sutures (thin dotted lines) are placed only in the connective tissue near the right and left hepatic arteries or the hepatoduodenal ligament at the porta hepatis. (B) Kasai’s original portoenterostomy:23,25 A continuous suture (looped line) is placed in the side of the transected biliary remnant, except at the 2 and 10 o’clock positions, where sutures (dotted lines) are placed in the connective tissue. (C) Extended portoenterostomy: Deep interrupted sutures (bold broken lines) are placed in the liver parenchyma, even at the 2 and 10 o’clock positions. (From Nakamura H, Koga H, Wada M, et al. Reappraising the portoenterostomy procedure according to sound physiological/anatomic principles enhances postoperative jaundice clearance in biliary atresia. Pediatr Surg 2012;28:205; and From Yamataka A, Lane GJ, Cazares J. Laparoscopic surgery for biliary atresia and choledochal cyst. Sem Pediatr Surg 2012;21:201.)
EPE is the most widely used open approach for treating biliary atresia. The patient is placed supine on an operating table with the capability for intraoperative cholangiography. An extended right subcostal incision, dividing the muscle layers, is used to expose the inferior margin of the liver. After division of the falciform and triangular ligaments, the liver is delivered from the abdominal cavity. This maneuver provides an excellent operative field for dissection of the porta hepatis. Cholangiography is recommended to identify the anatomy (Fig. 43-6). The fundus of the gallbladder is mobilized from the liver bed, and a 4–6 French feeding tube is passed into the gallbladder through a small cholecystotomy incision. If bile is detected on aspiration from the gallbladder, a small amount of contrast material is injected. However, in most patients with biliary atresia, the lumen of the atrophic gall bladder is already obstructed, and it is impossible to insert even a 4 French catheter. Thus, cholangiography cannot be performed. Unless normal anatomy of the intrahepatic biliary system can be confirmed, hepatic portoenterostomy is performed.24,82
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


