Urea Cycle and Related Disorders
Mendel Tuchman, Uta Lichter-Konecki, and Mark L. Batshaw
Ammonia is a degradation product of nitrogen-containing compounds (mainly proteins and amino acids) and is generated by the metabolism in all living organisms. Dietary protein contains approximately 16% nitrogen, and the excess nitrogen from the amino acids that are not incorporated back into proteins is excreted in the urine as urea. Urea is produced in the liver by an enzymatic process that is dependent on an intact urea cycle. When the urea cycle malfunctions, ammonia accumulates in blood and tissues.
The physiology of ammonia production and disposal is summarized in Figure 145-1. The normal level of free ammonia in plasma of humans is usually less than 35 μmol/L. Levels less than 50 μmol/L are considered safe while levels above 100 μmol/L are frequently associated with a range of clinical symptoms, including lethargy, vomiting, and altered consciousness. The severity of symptoms usually correlates with the degree of hyperammonemia, but this correlation may vary in different patients—while some patients may show severe brain dysfunction at ammonia levels between 100 and 200 μmol/L, others may function normally at a similar degree of hyperammonemia. 
The exact mechanism(s) by which ammonia exerts its toxicity on the brain is not well understood, although research points to several potential toxic effects. 
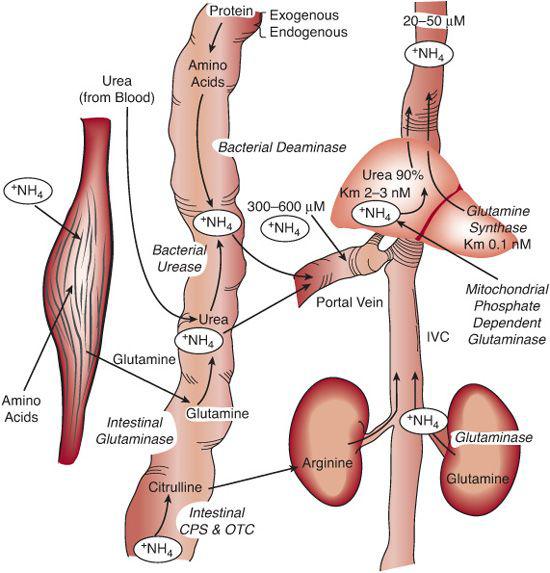
FIGURE 145-1. Human ammonia physiology. The complete urea cycle resides exclusively in the liver. The intestine is a source of ammonia and citrulline. Most of the ammonia that is absorbed from the intestine is converted to urea in the liver. Ammonia concentration in the portal blood is approximately 10 times higher than in systemic blood. The citrul-line produced in the intestine bypasses the liver and is converted to arginine in the kidney. Most circulating glutamine originates in the muscle, and smaller amounts come from the liver. CPS, carbamyl phosphate synthetase; OTC, ornithine transcarbamylase.
Hyperammonemia causes swelling of astrocytes, microglia cells that express the enzyme glutamine synthetase. Essentially all ammonia that enters the brain is incorporated into glutamine, so it is possible that glutamine, rather than ammonia, is the primary toxic metabolite in hyperammonemia.  Clinically, the effect of ammonia toxicity results in brain edema, which in severe cases leads to vascular compromise or brain herniation and death.
Clinically, the effect of ammonia toxicity results in brain edema, which in severe cases leads to vascular compromise or brain herniation and death.
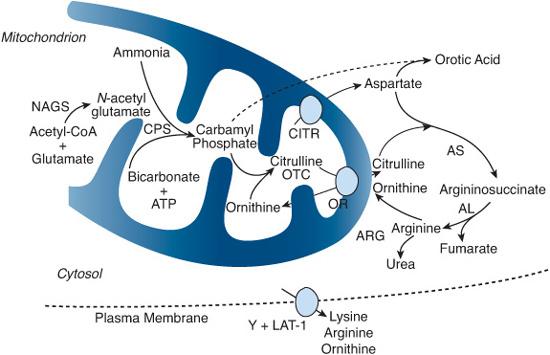
FIGURE 145-2. The urea cycle consists of six enzymes and two mitochondrial membrane transporters. An additional plasma membrane transporter is also important for urea formation. Each of these components is labeled in red letters and is deficient in one of the disorders discussed in this chapter. NAGS, N-acetylglutamate synthase; CPS, carbamyl phosphate synthetase; OTC, ornithine transcarbamylase; AS, argininosuccinate synthetase (citrullinemia); AL, argininosuccinate lyase (argininosuccinic aciduria); ARG, arginase (argininemia); ORNT, ornithine transporter (hyperornithinemia, hyperammonemia, and homocitrullinuria); CITR, citrin (glutamate/aspartate transporter, citrullinemia type II); Y+LAT-1, dibasic amino acid transporter (hyperdibasic aminoaciduria or lysinuric protein intolerance [LPI])
The complete urea cycle resides exclusively in periportal hepatocytes and is an essential biochemical pathway for waste nitrogen excretion.  Nine proteins are currently known to be directly involved in ureagenesis (Fig. 145-2).1 They consist of six enzymes and three membrane transporters. Three enzymes (proximal) are located within the mitochondrial matrix: N-acetylglutamate synthase (NAGS), carbamyl phosphate synthetase I (CPS), and ornithine transcarbamylase (OTC). The remaining three enzymes (distal)—argininosuccinate synthetase (AS), argininosuccinate lyase (AL), and arginase (ARG)—are located in the cytosol. Ornithine translocase (ORNT) and citrin (CITR), two transporters located in the inner mitochondrial membrane, allow the shuttling of amino acid intermediates (ornithine and aspartate, respectively) between the two cellular compartments. In addition, in the neonatal period, ornithine is provided to the urea cycle from the transamination of pyrroline-5-carboxylate (P5C) by ornithine amino transferase, which is oriented transiently toward the ornithine synthesis (see Chapter 161).
Nine proteins are currently known to be directly involved in ureagenesis (Fig. 145-2).1 They consist of six enzymes and three membrane transporters. Three enzymes (proximal) are located within the mitochondrial matrix: N-acetylglutamate synthase (NAGS), carbamyl phosphate synthetase I (CPS), and ornithine transcarbamylase (OTC). The remaining three enzymes (distal)—argininosuccinate synthetase (AS), argininosuccinate lyase (AL), and arginase (ARG)—are located in the cytosol. Ornithine translocase (ORNT) and citrin (CITR), two transporters located in the inner mitochondrial membrane, allow the shuttling of amino acid intermediates (ornithine and aspartate, respectively) between the two cellular compartments. In addition, in the neonatal period, ornithine is provided to the urea cycle from the transamination of pyrroline-5-carboxylate (P5C) by ornithine amino transferase, which is oriented transiently toward the ornithine synthesis (see Chapter 161).
A ninth protein (Y+LAT-1) involved in the transport of dibasic amino acids (lysine ornithine and arginine) across the cell’s plasma membrane is also important for normal function of the urea cycle (see Chapter 162).
A molecule of free ammonia interacts with carbon dioxide and ATP to produce carbamyl phosphate, catalyzed by carbamyl phosphate synthetase I. This enzyme requires N-acetylglutamate (NAG), a cofactor that is produced within the mitochondria by N-acetylglutamate synthase. Ornithine transcarbamylase then catalyzes the interaction between carbamyl phosphate and ornithine to form citrulline. Citrulline is transported from the mitochondria into the cytosol by ornithine translocase and interacts there with aspartate (which is transported by citrin) to form argininosuccinate. Aspartate contributes the second nitrogen atom (the first coming from free ammonia). Argininosuccinate synthetase is the enzyme that catalyzes this reaction. Argininosuccinate is hydrolyzed to arginine and fumarate by argininosuccinate lyase, and arginine is split into urea and ornithine by arginase. Urea is transported into the blood and eliminated in the urine while ornithine is transported into the mitochondria by ornithine translocase to resume the cycle. Thus, in every turn of the urea cycle, one molecule of urea is produced from free ammonia, the nitrogen of aspartate, and the carbon of carbon dioxide. 
Ammonia is also taken up by natural “scavengers” (eg, glutamate, pyruvate, and aspartate) and is used in the synthesis of nitrogen-containing compounds (eg, glycine and pyrimidines, including orotic acid).
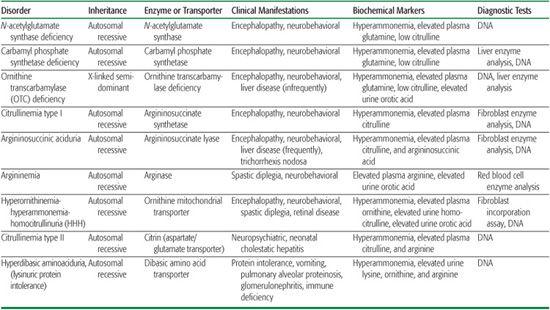
Urea cycle disorders are caused by specific defects in any of the eight genes encoding enzymes or membrane transporters involved in ureagenesis (Fig. 145-2). Their overall prevalence is estimated at 1:8,200. Urea cycle disorders, listed in Table 145-1, should be distinguished from other inborn errors of metabolism with hyperammonemia, including lysinuric protein intolerance (Chapter 162), organic acidemias (Chapter 156), fatty acid oxidation disorders (Chapter 170), and energy metabolism disorders (Chapter 176). Congenital disorders must also be differentiated from the acquired hyperammonemia seen in liver disease of various causes and following chemotherapy and organ transplantation. Finally, there is a rare and severe condition termed transient hyperammonemia of the neonate that is found predominantly in premature infants, the etiology of which remains obscure.
 BIOCHEMICAL BASIS OF UREA CYCLE DISORDERS1
BIOCHEMICAL BASIS OF UREA CYCLE DISORDERS1
Inherited disorders of the urea cycle could be classified as (1) proximal defects: N-acetylglutamate synthase, carbamyl phosphate synthetase I, and ornithine transcarbamylase deficiency; (2) distal defects: argininosuccinate synthetase deficiency (citrullinemia), argininosuccinate lyase deficiency (argininosuccinic aciduria), and arginase deficiency (argininemia or hyperargininemia); and (3) membrane transport defects: ornithine translocase deficiency (hyperornithinemia, hyperammonemia, and homocitrullinuria or HHH; see Chapter 161), citrin deficiency (citrullinemia type II), and dibasic amino acid transporter deficiency (hyperdibasic aminoaciduria or lysinuric protein intolerance [LPI]; see Chapter 162). Awareness of these disorders is important, because failure to recognize hyperammonemia often leads to brain damage or death  .
.
A functional block of the urea cycle results either from an enzyme deficiency (N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate synthetase, argininosuccinate lyase, and arginase) or from depletion of an amino acid that is essential to the cycle’s normal function and results from a transport defect (HHH, citrullinemia type II, LPI, and OAT in the neonatal period). 
Expanded newborn mass screening is now available for citrullinemia, argininosuccinic aciduria, and possibly argininemia and HHH syndrome. 
 CLINICAL PRESENTATION2-6
CLINICAL PRESENTATION2-6 
The most severe cases lack enzymatic activity and usually present with acute hyperammonemic coma within the first week of life, while patients with the milder forms have some residual enzyme activity, and their clinical presentation occurs later in life (from infancy through adulthood) with recurrent episodes of hyperammonemia.
Table 145-1. Urea Cycle and Related Disorders
Approximately 15% of OTC-deficient heterozygotes develop symptoms of hyperammonemia sometime in their lives, presumably as a result of skewed inactivation (lionization) of the normal OTC allele. Conversely, occasional asymptomatic adults have been found to harbor the same genetic defect that is causing symptoms in a family relative.
NEONATAL-ONSET UREA CYCLE DISORDERS
Infants with complete enzyme defects are usually born at term with normal Apgar scores.  However, between 1 and 5 days of age, they start feeding poorly, vomit frequently, gradually become lethargic and hypotonic, and may hyperventilate. The diagnosis of sepsis is frequently considered. These patients progressively develop tremor, stupor, apnea, coma, increased intracranial pressure, and death if the hyperammonemia is not diagnosed and treated effectively. Plasma ammonia levels can rise to extremely high levels, frequently above 1000 μmol/L (normal < 35). Initial blood-gas measurement typically shows a respiratory alkalosis, which is unique to hyperammonemia, because in most cases of similar clinical presentation due to other causes, including sepsis, a metabolic acidosis is present. Pulmonary bleeding has been reported as a terminal event, but more frequently the cause of death is vascular compromise and herniation of the brain.
However, between 1 and 5 days of age, they start feeding poorly, vomit frequently, gradually become lethargic and hypotonic, and may hyperventilate. The diagnosis of sepsis is frequently considered. These patients progressively develop tremor, stupor, apnea, coma, increased intracranial pressure, and death if the hyperammonemia is not diagnosed and treated effectively. Plasma ammonia levels can rise to extremely high levels, frequently above 1000 μmol/L (normal < 35). Initial blood-gas measurement typically shows a respiratory alkalosis, which is unique to hyperammonemia, because in most cases of similar clinical presentation due to other causes, including sepsis, a metabolic acidosis is present. Pulmonary bleeding has been reported as a terminal event, but more frequently the cause of death is vascular compromise and herniation of the brain.
LATE-ONSET UREA CYCLE DISORDERS
In patients with partial enzyme deficiencies, the first recognized clinical episode may be delayed for months or years; the symptoms are more subtle, and the hyperammonemia is generally less severe but can reach high levels if not promptly recognized and treated. The clinical abnormalities vary somewhat with the specific disorder. In most urea cycle disorders, the hyper-ammonemic episodes are marked by loss of appetite, cyclical vomiting, migrainelike episodes, ataxia, lethargy or hyperactivity, and behavioral abnormalities. Sleep disorders, delusions, hallucinations, and psychosis have also been reported. In adult males, some cases can mimic encephalitis, leading to death if hyperammonemia is not recognized and treated as an emergency.7 An encephalopathic (slow wave) EEG pattern may be observed during hyperammonemia; white matter lesions and nonspecific brain atrophy may be observed by MRI.
 ASSOCIATED CLINICAL ABNORMALITIES
ASSOCIATED CLINICAL ABNORMALITIES
In addition to hyperammonemia symptoms, several hyperammonemic disorders have other more specific clinical abnormalities. In hyper-argininemia, there is a progressive spastic diplegia or quadriplegia that has also been observed in HHH and can be the presenting sign. Tremor, ataxia, and choreoathetosis have been reported in hyperargininemia as well, whereas retinal de-pigmentation and chorioretinal thinning have been observed in HHH (see Chapter 142). Interstitial pneumonia due to pulmonary alveolar proteinosis is seen in LPI, as is glomerulonephritis and osteoporosis. There may also be an underlying immune deficiency in this disorder (see Chapter 143). Trichorrhexis nodosa, a nodelike appearance of fragile hair, is pathognomonic for argininosuccinic aciduria. Patients with this disorder can also develop liver fibrosis or cirrhosis.
In the neonatal period, citrullinemia type II can have an unusual presentation in the form of cholestatic hepatitis, which could be transient and reappear later in childhood or more commonly in adulthood with sleep disorder, recurrent vomiting and diarrhea, seizures, and behavioral and psychiatric symptoms. These patients have a spontaneous aversion for carbohydrates.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
The diagnosis of congenital hyperammonemia should be entertained upon finding an elevated plasma ammonia level in association with only mild or absent liver dysfunction, and in the absence of ketoacidosis. A precipitating catabolic event such as an infection, traumatic injury, ingestion of large amounts of protein, or other yet unknown metabolic stresses can precipitate hyperammonemia in these patients. Valproate or haloperidol administration has unmasked, un-diagnosed urea cycle defects in some patients, while other patients have erroneously been diagnosed as having Reye syndrome. 
The first measure to be taken is a plasma ammonia level. Blood should be as free flowing as possible while being collected, then placed on ice and analyzed within 30 minutes of collection. Most hospitals now have automated analyzers that measure ammonia and require less than 1 mL of blood with an analysis time of less than 30 minutes. In symptomatic hyperammonemia, ammonia levels are almost always higher than 100 μmol/L. Other routine laboratory tests may be useful in making the diagnosis. A low blood urea nitrogen concentration and respiratory alkalosis in a severely ill child are characteristic of urea cycle disorders. On the other hand, a metabolic acidosis or ketoacidosis is more commonly seen in hyperammonemia caused by an organic acidemia or congenital lactic acidosis.  Fatty acid oxidation defects can be distinguished by the low degree of ketosis and by specifically measuring acylcarnitine esters in blood (Chapter 170).
Fatty acid oxidation defects can be distinguished by the low degree of ketosis and by specifically measuring acylcarnitine esters in blood (Chapter 170).
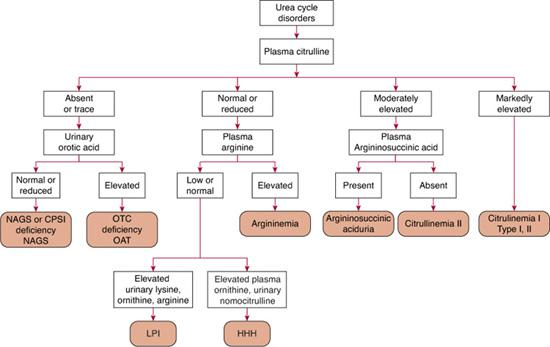
Quantitative plasma and urinary amino acids are most helpful in establishing a specific diagnosis of a defect in urea synthesis. Concentrations of glutamine, alanine, and asparagine, serving as storage forms of waste nitrogen, are typically increased. Plasma arginine concentration is decreased in all urea cycle disorders, except in argininemia, in which it is markedly increased. Plasma citrulline levels help discriminate between the proximal and distal urea cycle defects, because citrulline is the product of ornithine transcarbamylase (OTC) and carbamyl phosphate synthetase I (CPS) activity and is a substrate for the distal enzymes. As a result, no citrulline, or only a trace, is detected in the plasma in neonatal-onset CPS and OTC deficiencies; concentrations are low to low-normal in late-onset disease but are markedly elevated in citrullinemia and argininosuccinic aciduria. To distinguish CPS from OTC deficiency, urinary orotic acid is measured; concentrations are significantly elevated in OTC deficiency and are normal or low in CPS deficiency. In addition to OTC deficiency, urinary orotic acid excretion can be increased in argininemia, citrullinemia, HHH syndrome, and LPI. Ornithine amino transferase deficiency (OAT) can mimic OTC deficiency in neonates (hyperammonemia, low citrulline, orotic aciduria). Diagnosis is difficult, as hyper-ornithinemia—the gold marker of OAT—can be absent for the first few months of life.
In patients with citrullinemia type I, elevation in plasma citrulline levels can be up to a hundredfold, while those with citrullinemia type II (citrin deficiency) have only a moderate hypercitrullinemia. Argininosuccinic aciduria shows a more moderate increase in citrulline of about tenfold, associated with the presence of normally absent argininosuccinic acid.  In HHH syndrome, plasma ornithine and urine homocitrulline levels are elevated. In LPI, plasma levels of arginine, ornithine, and lysine are low, contrasting with extremely elevated urinary lysine, arginine, and ornithine excretion without concomitant elevated cystinuria. This profile is highly diagnostic (Chapter 162).
In HHH syndrome, plasma ornithine and urine homocitrulline levels are elevated. In LPI, plasma levels of arginine, ornithine, and lysine are low, contrasting with extremely elevated urinary lysine, arginine, and ornithine excretion without concomitant elevated cystinuria. This profile is highly diagnostic (Chapter 162).
Citrullinemia, argininosuccinic aciduria, argininemia, LPI, and HHH syndrome can be diagnosed on the basis of the amino acid pattern. Carbamyl phosphate synthetase I and OTC deficiency can be diagnosed by enzyme assay of a liver biopsy sample. Diagnosis by mutation analysis is now possible for all urea cycle disorders, and in OTC deficiency, 80% of patients have an identifiable mutation when the gene’s exons and their boundaries are screened.
GLUTAMINE SYNTHETASE DEFICIENCY
Inherited systemic deficiency of glutamine based on a defect of glutamine synthetase (GS) was recently described in two unrelated newborns with an early fatal course of the disease. Brain malformation (agyria), severe enteropathy, necrolytic erythema of the skin, severe hypoglutaminemia (<10 μmol/L; N >400) and a slight elevation of ammonia (110 μmol/L) were present. GS activity in transformed lymphocytes was absent, and a homozygote null mutation was found in both cases. These cases emphasize the role of GS in fetal development.8
 GENETICS
GENETICS
All described disorders except ornithine transcarbamylase (OTC) have autosomal recessive inheritance. DNA testing is available for all, allowing antenatal diagnosis and familial studies. Heterozygotes are asymptomatic. Plasma citrulline is slightly elevated (40 to 60 μmol/L) in heterozygotes for citrullinemia type I. OTC is an X-linked disorder. When the diagnosis of OTC is established, it is necessary to take a careful family history and to assess the mother’s carrier status, most reliably by mutation analysis. If the mutation is not known, the most convenient investigation is the allopurinol test, which is used to detect increased de novo pyrimidines synthesis (orotic acid excretion). This test is not entirely specific or sensitive.1 One should be aware of asymptomatic forms in hemizygote adult males with residual enzyme activity, as they can transmit the mutation to all their daughters.
 TREATMENT APPROACHES9-12
TREATMENT APPROACHES9-12
Treatment approaches rely on a combination of dietary nitrogen restriction, replacement of deficient amino acids, and stimulation of alternate pathways for waste nitrogen excretion. The protein-restricted diet is often supplemented with essential amino acids. Arginine is deficient in all disorders except argininemia, and it must be replaced. Hair fragility and an exematous rash are associated with arginine deficiency, which can develop if arginine replacement is inadequate. Alternate pathway therapy uses benzoate, which is conjugated in the liver with glycine to form hippuric acid, and phenylacetate (or its prodrug phenylbutyrate, Buphenyl), which is conjugated in the liver with glutamine to form phenylacetylglutamine. Both hippurate and phenylacetyl-glutamine are readily eliminated in the urine, thereby providing an alternate pathway for waste nitrogen excretion. In addition, arginine stimulates waste nitrogen excretion in citrullinemia and argininosuccinic aciduria.
 TREATMENT OF ACUTE HYPERAMMONEMIA IN NEWBORNS
TREATMENT OF ACUTE HYPERAMMONEMIA IN NEWBORNS
Acute hyperammonemia represents a medical emergency (Fig. 145-3). Studies have shown that hyperammonemic coma lasting longer than 72 hours invariably leads to severe brain damage and intellectual disability. When newborns are in a coma due to plasma ammonia levels being above 200 μmol/L, hemodialysis should be used if possible; if hemodialysis is not available, hemofiltration or peritoneal dialysis should be used.13 Protein intake is discontinued until ammonia levels come under control, and intravenous glucose and lipids are administered to decrease catabolism. In addition, alternative pathways are exploited to eliminate nitrogen by the intravenous administration of sodium benzoate and sodium phenylacetate (Ammonul). Arginine hydrochloride 10% solution (R-gene) is given to prevent arginine deficiency. Sterilization of the gut may also be useful in reducing ammonia absorption. Treatment of increased intracranial pressure is also very important. The comatose child should be placed on a ventilator with mild hyperventilation, and drugs should be administered to reduce intracranial pressure. 
 TREATMENT OF ACUTE HYPERAMMONEMIA IN OLDER CHILDREN
TREATMENT OF ACUTE HYPERAMMONEMIA IN OLDER CHILDREN
In children with partial (late-onset) defects, and in those with neonatal-onset disease who are on alternate pathway therapy, intercurrent hyper-ammonemic episodes may be treated initially in a more conservative manner than in the newborn period (Table 145-2, Fig. 145-4). If plasma ammonia levels are below 200 μmol/L and the child is not in a coma, intravenous treatment with benzoate, phenylacetate, and L-arginine can be tried initially. However, if the plasma ammonia level does not fall within hours or if the clinical condition worsens, dialysis should be started. To ameliorate catabolism, glucose and lipids should also be given.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


