Trypanosomiases
Eduardo Ortega-Barria
AFRICAN TRYPANOSOMIASIS
African trypanosomiasis (sleeping sickness) is a parasitic disease transmitted by tsetse flies of the genus Glossina and caused by a group of parasites called trypanosomes.1 West African trypanosomiasis, also called Gambian sleeping sickness, is caused by a subspecies of the extracellular flagellate Trypanosoma brucei known as Trypanosoma brucei gambiense. East African trypanosomiasis, also called Rhodesian sleeping sickness, is caused by T brucei rhodesiense. These diseases exist in Africa wherever the various species of Glossina (ie, the tsetse fly) are found.
 EPIDEMIOLOGY
EPIDEMIOLOGY
African trypanosomiasis has a focal distribution but expands outward during epidemics. About 60 million people live in risk areas of African trypanosomiasis, and the World Health Organization estimates a continent-wide prevalence of both forms of African trypanosomiasis of 300,000 cases; an average of 30,000 new cases of the disease have been reported annually over the last 10 years, although underreporting rates are high.1 Thirtysix sub-Saharan countries are considered endemic for one or the other form of the disease despite that some of them have reported no cases in the last decade.2 The Gambian form is currently a major public health problem over vast areas of Africa from Sudan in the north to northwest Uganda to the Democratic Republic of the Congo to Angola in the south. This form represents more than 90% of reported cases.2 The Rhodesian form continues to present a serious health risk in the Lake Victoria Basin, particularly in eastern Uganda, and there are small pockets of disease endemic in other countries of east and central Africa. A line drawn from north to south through sub-Saharan Africa, roughly following the Rift Valley, differentiates the distribution of the 2 diseases with the Gambian form to the west and the Rhodesian to the east of this notional line.1T brucei rhodesiense is a zoonotic parasite, and wild and domestic animals serve as reservoirs. A number of domestic animals species, including pigs, dogs, goats, sheep, and cattle, may carry human-infective trypanosome species. For T brucei gambiense, the nature of animal reservoirs and its role in disease transmission is somewhat less certain.1
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
All members of the T brucei complex share a common morphology, biochemistry, and life cycle (eFig. 355.1  ). Infective metacyclic trypomastigotes are inoculated into subcutaneous tissue of a human or another mammalian host by a bite from the tsetse fly. They are converted to the pleomorphic blood forms: long, slender forms (20–40 by 1 μm) and stumpy forms (15–25 by 3.5 μm). Within the human host, trypomastigotes multiply in blood, lymph, and extracellular spaces. The central nervous system eventually is invaded, where multiplication continues unabated. The tsetse fly is infected with ingestion of a blood meal. Trypomastigotes differentiate into procyclic forms in the midgut where they multiply by binary fission and remain for the next 2 to 3 weeks. Finally, they enter the salivary glands and transform into infective metacyclic trypomastigote forms. When the fly bites another mammal, the parasites are injected into the blood, completing the cycle of infection.
). Infective metacyclic trypomastigotes are inoculated into subcutaneous tissue of a human or another mammalian host by a bite from the tsetse fly. They are converted to the pleomorphic blood forms: long, slender forms (20–40 by 1 μm) and stumpy forms (15–25 by 3.5 μm). Within the human host, trypomastigotes multiply in blood, lymph, and extracellular spaces. The central nervous system eventually is invaded, where multiplication continues unabated. The tsetse fly is infected with ingestion of a blood meal. Trypomastigotes differentiate into procyclic forms in the midgut where they multiply by binary fission and remain for the next 2 to 3 weeks. Finally, they enter the salivary glands and transform into infective metacyclic trypomastigote forms. When the fly bites another mammal, the parasites are injected into the blood, completing the cycle of infection.
The inoculated metacyclic trypomastigotes transform and multiply locally in the subcutaneous tissue, giving origin to a characteristic hard and sometimes painful chancre. By about the 10th day, slender forms reach the bloodstream and lymphatics, and for the next several days, their numbers increase logarithmically, spreading to several tissues. Soon thereafter, the organisms nearly disappear from the bloodstream, only to reappear later. After a variable length of time, trypomastigotes pass through the blood-brain barrier and induce inflammation of the brain tissue (meningoencephalitis) as well as cellular infiltration of spinal nerve tissue.
The parasite survives in the mammalian host by periodically altering its surface antigenic coat, evading the developing immune response of the host. Each successive parasitemic wave represents a new antigenic variant that has emerged to elude the host’s antibody response to the previous antigen. The parasite is covered with a variable surface glycoprotein (VSG). Each peak of parasitemia contains a predominant variable antigen type (VAT). The specific antibody response to this coat leads to the destruction of the predominant VAT or homotype. However, within each population of parasites are a number of heterotypes, one of which then becomes the next homotype that is not recognized by the host’s immune response. The parasite in each successive wave of parasitemia bears a different VAT. A single trypomastigote may contain as many as 1000 genes, each encoding for a specific VSG.
A marked specific humoral antibody response (predominantly immunoglobulin M [IgM]) follows infection. As a result of polyclonal B-cell activation, there also are many antibodies produced to a wide variety of antigens, including red blood cells, brain, and heart. Circulating immune complexes have been reported regularly, and these may be responsible for the glomerulonephritis often accompanying acute and chronic disease.
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
Clinical manifestations with T brucei gambiense and T brucei rhodesiense are similar except that Rhodesian infection presents with a more acute and severe disease, with fewer central nervous system symptoms, and if untreated, death occurs within a few months. Gambian infection has a milder, chronic course, with involvement of lymph nodes, central nervous system invasion, and a fatal outcome after several years.
In both forms, the trypanosomal chancre may be evident 2 to 3 days after the bite of the fly (Fig. 355-1 and eFig. 355.2  ). Within 1 week, the lesion becomes a large, red, warm, and painful nodule that usually resolves within 3 weeks, leaving residual scarring and depigmentation. In Rhodesian trypanosomiasis, the incubation period typically is 2 to 3 weeks, whereas with Gambian infection, the onset of symptoms may be delayed for several weeks or years. Early stages are characterized by intermittent fevers, chills, headache, and generalized lymphadenopathy. The nodes are discrete, soft, nontender, and contain abundant parasites. In Gambian disease, nodes in the posterior cervical triangle may become enlarged (ie, Winter-bottom sign) and, when present, strongly suggest the diagnosis. The spleen and liver may be mildly enlarged. Intermittent fevers may last for months to years with Gambian infection. Wasting and malnutrition occur, and patients often die of intercurrent infections. An irregular, circinate, evanescent rash on the trunk, shoulders, and thighs that is more evident in white persons may appear. The sedimentation rate is elevated markedly; anemia, hypoalbuminemia, and thrombocytopenia are frequent as well. Serum immunoglobulin levels (IgM), circulating immune complexes, heterophil antibodies, and rheumatoid factor are increased.
). Within 1 week, the lesion becomes a large, red, warm, and painful nodule that usually resolves within 3 weeks, leaving residual scarring and depigmentation. In Rhodesian trypanosomiasis, the incubation period typically is 2 to 3 weeks, whereas with Gambian infection, the onset of symptoms may be delayed for several weeks or years. Early stages are characterized by intermittent fevers, chills, headache, and generalized lymphadenopathy. The nodes are discrete, soft, nontender, and contain abundant parasites. In Gambian disease, nodes in the posterior cervical triangle may become enlarged (ie, Winter-bottom sign) and, when present, strongly suggest the diagnosis. The spleen and liver may be mildly enlarged. Intermittent fevers may last for months to years with Gambian infection. Wasting and malnutrition occur, and patients often die of intercurrent infections. An irregular, circinate, evanescent rash on the trunk, shoulders, and thighs that is more evident in white persons may appear. The sedimentation rate is elevated markedly; anemia, hypoalbuminemia, and thrombocytopenia are frequent as well. Serum immunoglobulin levels (IgM), circulating immune complexes, heterophil antibodies, and rheumatoid factor are increased.
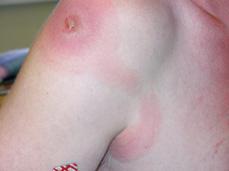
FIGURE 355-1. Trypanosomal chancre on shoulder with lymphangitis toward axilla. (From: http://www.cdc.gov/ncidod/EID/vol8no1/01-0130G1.htm.)
Untreated patients with Gambian disease often develop signs of central nervous system invasion. Severe headaches, insomnia, and a feeling of impending doom are typical. Next, there may be progressive mental deterioration, with patients becoming incapable of caring for themselves. Tremors, especially of the tongue, hands, and feet, and generalized or focal convulsions may occur. Myocarditis is particularly common and may lead to early death in the Rhodesian form of the disease. Untreated, both forms of sleeping sickness are always fatal.
The differential diagnosis includes tuberculosis, malaria, visceral leishmaniasis, brucellosis, relapsing fever, syphilis, enteric fever, and viral encephalitis.
 DIAGNOSIS
DIAGNOSIS
The diagnosis of human African trypanosomiasis is cumbersome and follows a 3-step approach: screening, parasitological confirmation (microscopic examination of lymph node and blood specimens), and staging (examination for trypanosomes and for increased white blood cell counts in the cerebrospinal fluid). Definitive diagnosis depends on demonstration of the parasite from the chancre, in blood, lymph node aspirates, bone marrow, or cerebrospinal fluid (CSF). Patent parasitemia is more common during febrile episodes. It is essential to use both thick and thin Giemsa-stained blood smears (Fig. 355-2) as well as to examine the buffy coat from 10 to 20 mL of citrated whole blood or the sediment from 5 mL of centrifuged cerebrospinal fluid. Concentration techniques such as the minihematocrit centrifugation technique or the miniature anion exchange centrifugation (m-AECT) improve detection levels to 500 and 100 parasites per milliliter by microscopy respectively. Because of the periodicity of the parasitemia, daily examination of blood films for 10 consecutive days is recommended. T brucei rhodesiense is found more frequently in peripheral blood and can be shown in a single microscopic examination in 87% of cases. Examination of wet preparations of cervical lymph node aspirates help to confirm the diagnosis when trypanosomes are undetected in blood. Intraperitoneal inoculation of 0.5 mL heparinized blood or CSF into 2 mice is positive in 89% of T brucei rhodesiense cases within 2 weeks of inoculation.
Diagnosis is followed by systematic stage determination, which consists of assessing the cerebrospinal fluid. In advanced central nervous system disease, the CSF shows increased protein concentration; elevated cell counts, predominantly mononuclear; small number of eosinophils; and, frequently, trypomastigotes. In advanced untreated sleeping sickness, the IgM level in the CSF often is elevated, but it has no relationship to the presence of trypanosomes in the CSF. After successful treatment, the IgM level declines gradually, disappearing after approximately 1 year.
Immunodiagnostic methods may be helpful for epidemiologic studies and detection of latent infections. Specific antibodies can be detected after the second week of infection by indirect fluorescent antibody or enzyme immunoassay. The card agglutination test, a simple, direct agglutination test for stained trypanosomes, available for the diagnosis of T brucei gambiense, has a specificity of 90% to 95% and a sensitivity of 92% to 100%.1 The test cannot differentiate between past and current infections and therefore cannot be used to assess treatment efficacy. Anther serologic test for T brucei gambiense infection is the latex/T brucei gambiense, which is a variant of the card agglutination test for T brucei gambiense but is directed against 3 variable antigen types and has a specificity of up to 99% and a sensitivity between 84% and 100%. Latex IgM can be used to detect elevated IgM in the central nervous system of second-stage patients: when T brucei gambiense trypanosomes invade the central nervous system.3
A monoclonal antibody–based enzyme immunoassay for detection of trypanosome antigens in serum and cerebrospinal fluid has a high specificity (99.9%) and sensitivity (94.8% rhodesiense). Finally, the polymerase chain reaction has been used to assess the presence of T brucei gambiense infection in both blood and cerebrospinal fluid of patients with a sensitivity of 100% in parasitologically positive samples and is recognized as being the most sensitive diagnostic method for African trypanosomiasis when compared with parasito-logical means.1
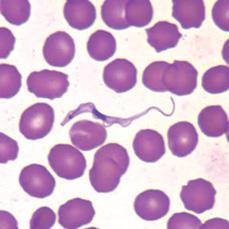
FIGURE 355-2. Trypanosoma brucei sp. in thin blood smear stained with Wright-Giemsa. (From: CDC: http://www.dpd.cdc.gov/dpdx/HTML/ImageLibrary/TrypanosomiasisAfrican_il.htm.)
 TREATMENT
TREATMENT
The choice of treatment depends on the parasite species involved and on the stage of the disease; drugs for the treatment of late-stage disease are able to cross the blood-brain barrier in order to clear trypanosomes in the cerebrospinal fluid. Four licensed compounds are used against human African trypanosomiasis today, depending on the causative subspecies and whether parasites have initiated disease of the central nervous system (stage 2) or not (stage 1).4-6 Two compounds are used against stage 1 disease: suramin and pentamidine. Against stage 2 disease, melarsoprol (active against T brucei gambiense and T brucei rhode-siense) and eflornithine (only useful against T brucei gambiense) can be used. Hospitalization is required during administration of all the drugs currently used, particularly for the late stage. Nifurtimox, alone or in combination with other drugs, particularly with eflornithine, is being considered as an option for melarsoprol-refractory late-stage disease.7,8
Early treatment is essential, when the parasite is restricted to the hematolymphatic system, because the prognosis is poor once central nervous system involvement has occurred. Suramin and pentamidine do not penetrate the central nervous system adequately and are useful only in early African trypanosome infections. Pentami-dine, as the isethionate salt, is used only against T brucei gambiense, as primary resistance of T brucei rhodesiense has been observed. Pentamidine is administered intramuscularly at 4 mg/kg/d for 7 to 10 doses, given daily or every second day. Both the liver and kidneys store the drug for months. Common adverse events include lowering of blood glucose levels, and hypotension is evident in 10% of treated cases. Other serious side effects include nephrotoxicity, hepatotoxicity, and pancreatotoxicity. Suramin, a polysulfonated naphthylamide, inhibits glycolytic enzymes of trypanosomes, and a full course cures early cases of T brucei gambiense and T brucei rhodesiense. Poor intestinal absorption and a local irritation if given intramuscularly mean that suramin is best given by slow intravenous injection. A test dose of 5 mg/kg is given and is followed by a dose of 20 mg/kg intravenously (maximum dose 1 g) on days 1, 3, 7, 14, and 21. Common reactions include joint pain, fever, and paresthesia. Neuropathy, rash, fatigue, anemia, hyperglycemia, hypocalcemia, coagulopathies, neutropenia, renal insufficiency, and transaminases elevation are all common.6 Nephrotoxicity is the most important toxic effect; urinalysis should be done prior to administration of each dose, and the presence of casts or red blood cells in the sediment are indications for alternative therapy.
During the second stage of the disease, drugs that can penetrate into the cerebrospinal fluid are indispensable. Only the arsenical compound melarsoprol has demonstrated the ability to cross the blood-brain barrier and kill T brucei gambiense and T brucei rhodesiense parasites residing in the cerebrospinal fluid,4 although melarsoprol maximally accumulates across the blood-brain barrier to levels only around 1% to 2% of maximum plasma levels. Various empirically designed, complicated treatment schedules are used. All consist of 3 or 4 series of 3 to 4 injections, with intervening rest periods of 7 to 10 days. One standard schedule uses 3 series of 4 daily injections of melarsoprol at doses increasing from 1.2 to 3.6 mg/kg within each series, with a 7-day interval between series.9 A new standardized 10-day course with melarsoprol at 2.2 mg/kg given once a day is as effective as older protocols and has been adopted as the recommended course for T brucei gambiense by World Health Organization.9,10 Treatment failures with melarsoprol have been reported in some regions to reach 30%.4,6 The most serious side effect is encephalopathy, which can occur in 1% to 10% of treated patients, with mortality in 1% to 5%. Other adverse events are common. These include cutaneous reactions, polyneuropathy, fever, headache, pruritus, and thrombocytopenia. Heart failure also has been reported. Coadministration of corticosteroids yields some protection against the reactive encephalopathy. Thiamine administration can reportedly ameliorate melarsoprolís adverse event. Difluoromethylornithine is a selective and irreversible inhibitor of ornithine decarboxylase, the first enzyme in polyamine synthesis of trypanosomes. Recent trials indicate that in both the hemolymphatic and central nervous system stages of T brucei gambiense infection, difluoromethylornithine is effective and reliable; it is recommended for second-stage T brucei gambiense.11 The recommended dosage is 400 mg/kg/d intravenously in 4 divided doses for 14 days, although a higher dose is suggested for children and teenagers. Side effects include fever, headache, hypertension, macular rash, peripheral neuropathy, tremor, and gastrointestinal problems including diarrhea. A Jarisch-Herxheimer-type reaction has been reported. Nifurtimox, a 5-nitrofuran derivative, is active in both stages of T brucei gambiense infection.4 Success as monotherapy has reportedly been limited (50–80% cure), but its use, particularly in combinations, is of increasing interest. Recent clinical trials using eflornithine (400 mg/kg/day) for 7 days in 2 periods of infusion (rather than 14 days with 4 infusions) and nifurtimox at 15 mg/kg for 10 days have yielded cure rates as high as 98%.7,8
Control programs recommend a follow-up of patients with human African trypanosomiasis for 2 years after treatment, with examinations every 6 months for the presence of trypanosomes and assessment of the white blood cell (WBC) count in cerebrospinal fluid (CSF). A patient is declared to be cured if no trypanosomes are detected and the WBC count has returned to normal. Recently, it has been published that 6 months after treatment, patients with CSF WBC counts of less than 5 cells/μL were at low risk of human African trypanosomiasis recurrence (negative predictive value, > 0.93). After 12 months, the combination of CSF WBC count greater than 8 cells/μL and latex/IgM end titer greater than 1:4 predicted treatment failure with 97% specificity and 79% sensitivity. This same combination was 100% specific for treatment failure after 18 and 24 months.12
 PREVENTION
PREVENTION
The control of trypanosomiasis is concerned with reducing parasite transmission from host to vector and on to further hosts.1 For the control of Gambian African trypanosomiasis, combining active case detection and drug treatment of positive cases is now the cornerstone of disease prevention and control. Reducing the size of the human reservoir will reduce the chances of further generations of tsetse becoming infected and passing on the infection, so that transmission will eventually be interrupted. While finding and treating infected patients is of course a necessary ethical duty and medical task, such activities will not stem the actual transmission of T brucei rhodesiense parasites. Rather, livestock and other animal hosts of this zoonotic subspecies need to be the focus of control activities.1 Vector control can curb transmission and has been successfully employed in recent years to eradicate trypanosomiasis using extensive trapping, followed by the sterile insect technique. Tsetse flies can be controlled by using insecticides, such as chlorinated hydrocarbons and synthetic pyrethroids directed at known fly resting sites. Fly traps or screens impregnated with insecticides may be more economical and effective. Chemoprophylaxis with suramin, 0.3 to 0.7 g intravenously every 2 to 3 months, is reported to be highly effective, although it is no longer used. A process of antigenic variation, whereby parasites repeatedly change the surface coat that interfaces with the immune system, renders the prospects of vaccination poor.
AMERICAN TRYPANOSOMIASIS
American trypanosomiasis (ie, Chagas disease) is caused by the obligate intracellular protozoan parasite Trypanosoma cruzi and threatens more than 120 million (ie, 25%) of the population of Latin America. Transmission usually occurs by vector transfer, by transfusion, or by congenital transmission.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Human Chagas disease extends from the southern United States to Chile and Argentina. The Caribbean, Belize, Surinam, and Guyana are reported to be free of this infection. Approximately 16 million to 18 million people are infected in Latin America, with 200,000 new cases occurring a year. The prevalence of infection is greatest in Brazil, Argentina, Bolivia, and Venezuela. There may be up to 100,000 immigrants from endemic countries with latent T cruzi infections living in the United States and representing a reservoir for potential transmission of T cruzi by blood transfusion or organ donation. The distribution of Chagas disease in the United States includes approximately the southern half of the country; the disease exists almost exclusively as a zoonosis, and only 5 autochthonous cases of Chagas disease have been documented.14 Despite this apparent low incidence, there is evidence of a relatively high rate of infection among recognized indigenous triatomid bugs in southern states; it has been suggested that Chagas disease is endemic to Texas and that unrecognized cases exist throughout the state. On the other hand, several cases acquired through laboratory accidents, organ transplants, imported infection, and blood transfusion have been reported in the United States and Canada.15-17
T cruzi infection can be readily transmitted by blood transfusion (20% infectivity risk). The migration of people infected by T cruzi poses a threat to those countries where this insect-transmitted disease does not occur. From 2001 to 2002, the prevalence rate of serologic markers in blood donors ranged from 99 per 1000 in Bolivia to 1.5 per 1000 in Ecuador.18 Estimates of T cruzi seroprevalence in US blood donors range from 0.01% to 0.20% and are higher in geographic regions with higher rates of Hispanic donors. A large multiyear seroprevalence study of T cruzi in US blood donors reported an overall seropositive rate for all donations in Los Angeles and Miami to be 1 in 7,500 and 1 in 9,000 donations respectively.
Congenital transmission occurs in Latin American countries where Chagas disease is prevalent in women of reproductive age with chronic T cruzi infection. Congenital transmission ranges from 1% or less in Brazil to 7% or more in some regions of Bolivia, Chile, and Paraguay. Finally, in endemic areas, transmission has been associated with the ingestion of contaminated food or drink.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The disease is transmitted by bloodsucking insects that become infected by ingesting trypomastigotes present in the bloodstream of infected mammals. The organisms reach the insect midgut, transform into the multiplying epimastigote stage, and change progressively to infective metacyclic trypomastigotes until they are excreted with feces and urine during feeding. These enter the human body through an abrasion on the skin, intact mucous membrane, or the bite wound.13
Two theories have been set forth to explain the pathogenesis of the lesions in Chagas disease. The parasite persistence theory postulates that Chagas lesions could be a direct consequence of the mechanical rupture of the parasitized host cell and subsequent inflammation.19 The autoimmune theory ascribes Chagas lesions to the rejection of parasite-free target cells by immune lymphocytes. Recently, horizontal DNA transfer from the parasite to the mammalian host has been implicated in the autoimmune response and pathogenesis of the disease.
The vectors of T cruzi are insects belonging to the order Hemiptera, family Reduviidae, and subfamily Triatominae. Currently, over 130 species are known, belonging to 5 tribes and 16 genera. However, only a few species of 3 genera—Triatoma, Rhodnius, and Panstrongylus—are important vectors of T cruzi between domestic animals and humans in endemic areas. All 3 genera are widely distributed in the Americas, from Mexico to Argentina and Chile. Twelve species of triatomines are known to occur in the United States, the most important being T sanguisuga in the eastern United States, T gerstaeckeri in the region of Texas and New Mexico, and T rubida and T protracta in Arizona and California. Human Chagas disease develops when humans come into contact with the natural foci of infection and disturb the environment, with the result that infected triatomines move into and colonize human dwellings.
In the mammalian host, the life cycle of T cruzi includes 2 developmental stages. The trypomastigote is found in the tissue and bloodstream of infected mammals and is responsible for the intercellular spreading of the infection. The trypomastigote is spindle shaped, measures about 20 μm, and has a kinetoplast located posterior to the nucleus and a flagellum that extends along the outer edge and reaches the anterior end of the body. In stained blood smears, trypomastigotes assume a characteristic C or S shape, with the nucleus just anterior to the middle of the cell (Fig. 355-3). The amastigote is the dividing intracellular form found in the tissues of the mammalian host. Once the cell fills with amastigotes, the amastigotes differentiate into mobile trypomastigotes that burst out, reach the bloodstream, and infect other cells of the body. Every cell in the human body with the exception of neurons and red blood cells could be affected, after which T cruzi infection persists in the human body for life. T cruzi isolates are grouped in lineages I and II, with type II divided into 5 subtypes, IIa through IIe. In South America, type II is predominantly distributed in Argentina, Chile, Paraguay, and Uruguay; whereas type I is associated with human disease north of the Amazon basin. In Mexico and the United States, most human cases are linked to type I. Lineage I is closely linked to the sylvatic cycle and apparently results in milder infections and morbidity in humans. Type II is closely related to the domestic cycle and produces major infections and morbidity in humans.
Natural infection occurs in a wide variety of peridomestic and sylvatic animals, including birds, guinea pigs, monkeys, opossums, foxes, ferrets, squirrels, armadillos, anteaters, porcupines, rats, and mice. Domestic dogs and cats are believed to be major reservoirs for human infection. However, within the domicile, transmission from human to human through the vector is probably the most common cycle of infection.
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
Chagas disease is divided into 3 distinct phases: acute, latent, and chronic. In 95% of patients, clinical symptoms during the acute phase are either mild or absent.13 Shortly after infection, parasites elicit a local inflammatory reaction at the site of entry that gives origin to the chagoma (nodular skin reaction) or the Romaña sign (unilateral periorbital edema, conjunctivitis, and preauricular lymphadenitis). Acute manifestations are usually seen in young children. Illness begins with fever, headache, anorexia, and lassitude. Cervical, axillary, and iliac lymphadenopathy; hepatosplenomegaly; vomiting; and rash are common, and occasionally, meningoencephalitis occurs.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


