Toxic Ingestions and Exposures
Sing-Yi Feng, Collin S. Goto, and M. Douglas Baker
MANAGEMENT OF THE INTOXICATED PATIENT
Morbidity and mortality from childhood poisoning have decreased in the last few decades. This decrease can be credited to the development of poison control centers with a sophisticated poison management database, new governmental regulations, widespread use of child-resistant enclosures for medications, safer packaging for consumer products, public education and anticipatory guidance, and a growing understanding of the environmental and pharmacologic foundations of toxicology.
In particular, poison control centers provide immediate and expert advice from trained specialists in poison information to aid the practitioner in the management of poisoned patients. In the United States, 1-800-222-1222 is the nationwide toll-free number connecting any caller to an American Association of Poison Control Center regional poison center. A clinical toxicologist is available on request to provide consultation regarding decontamination, antidotes or other medical treatment, selection of appropriate laboratory tests, and enhanced drug elimination.
POISON PREVENTION
The goal of poison prevention programs is to prevent pediatric poisoning through legislation and educational strategies such as parental anticipatory guidance during well-child visits (Table 120-1). Brochures and other poison prevention materials can be obtained from local poison centers or the American Academy of Pediatrics. Legislative initiatives such as the Poison Prevention Packaging Act have been instrumental in decreasing the impact of childhood poisoning.
EPIDEMIOLOGY
Young children usually have little difficulty finding toxic substances. Personalcare products and cleaning substances are the most common agents involved in household intoxications, though pharmaceutical products are responsible for the majority of fatalities. Analgesics are the most common pharmaceutical exposure.
There are fewer adolescent exposures reported to poison centers compared with younger children. Most adolescent poisonings are intentional, and involve greater amounts of toxin, as well as exposure to multiple agents. Exposures in the younger child are usually unintentional. Delayed presentation for medical attention can also complicate medical care. As a consequence, adolescent poisonings often result in more serious toxicity.
EVALUATION OF THE POISONED PATIENT
Determining the type of toxin involved guides therapy. The presumptive diagnosis can often be made using information from the history, vital signs, and physical examination before extensive laboratory results are available.
The type of toxin, timing, amount, and route of exposure are ascertained by asking the patient, family members, or other sources. The circumstances of the exposure and history of psychiatric illness help determine the risk for intentional harm. Potentially toxic household products such as automotive and cleaning products should not be overlooked. Paramedics provide important information about the scene of exposure and progression of the patient’s signs and symptoms. The poison center can aid in the identification of unknown pills.
 TOXIDROMES
TOXIDROMES
Toxidromes or “toxic syndromes” are characteristic vital signs and physical findings that indicate the presence of a specific category of toxins (Table 120-2). Toxidromes are especially helpful when the patient has been exposed to a single toxin. Exposure to multiple substances can obscure the clinical diagnosis, whereas other disease processes may mimic the toxidromes. Stimulation or inhibition of specific receptors in the autonomic and somatic nervous systems results in the classic toxidromes (eFig. 120.1  ). The opioid toxidrome results from stimulation of opiate receptors in the central nervous system. Withdrawal syndromes are associated with sudden abstinence after chronic use of sedative-hypnotic drugs or opioids.
). The opioid toxidrome results from stimulation of opiate receptors in the central nervous system. Withdrawal syndromes are associated with sudden abstinence after chronic use of sedative-hypnotic drugs or opioids.
 LABORATORY EVALUATION
LABORATORY EVALUATION
Basic laboratory evaluation provides valuable information for the diagnosis and management of the poisoned patient. An arterial blood gas demonstrating an acid–base disturbance can narrow the differential diagnosis. The electrolyte values will determine if there is an elevated anion gap metabolic acidosis suggesting the presence of a specific type of toxin (Table 120-3). The anion gap is estimated using serum mEq/L values:
Table 120-1. Poison Prevention Strategies
Install child-proof locks on cabinets and drawers that contain dangerous materials. |
Keep all medicines in child-proof containers and placed in a locked cabinet. |
Discard old medications. |
Keep all medications and chemicals in labeled containers. |
Keep alcohol and cigarettes away from the reach of children. |
Remove poisonous plants from the home and yard. |
Ask visitors and relatives to place medications where the child cannot reach them and not in a purse or suitcase. |
Keep the numbers of the local poison control center and emergency department by the telephone. |
[Na+] − ([Cl−] + [HCO3−])
where the bracketed symbols represent the serum concentrations of sodium, chloride and bicarbonate. An anion gap of 8 to 12 mEq/L is considered normal. An elevated anion gap suggests the presence of an unidentified negatively charged molecule in the serum, which may represent a toxin or a byproduct of the intoxication, such as lactate.
If there is an unexplained metabolic acidosis, obtaining serum osmolarity, electrolytes, blood urea nitrogen (BUN), and glucose and then calculating the osmolar gap can aid in diagnosis. An elevated anion gap metabolic acidosis accompanied by an elevated osmolar gap suggests toxic alcohol poisoning. The osmolar gap is calculated as:
Measured serum osmolality – Calculated serum osmolality
The measured serum osmolality is determined on a sample sent to the laboratory. The calculated osmolality can be estimated by the following formula:
2[Na] + [Glucose]/18 + [BUN]/2.8
An osmolar gap of 10 is considered normal. An elevated osmolar gap indicates the presence of a low-molecular-weight and osmotically active substance such as methanol or ethylene glycol in the blood. A normal osmolar gap does not eliminate the possibility of toxic alcohol poisoning if the alcohol has already been converted into toxic metabolites.
Quantitative blood tests are ordered for specific drugs or toxins whose serum concentration can guide therapy or predict toxicity. In cases of unknown ingestions, serum acetaminophen and salicylate levels are obtained because these poisonings are common and early diagnostic clues may be absent.
Table 120-2. Common Toxidromes
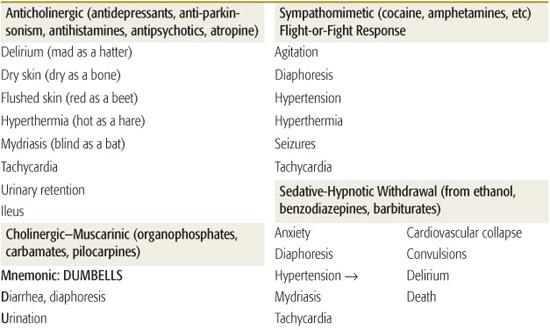
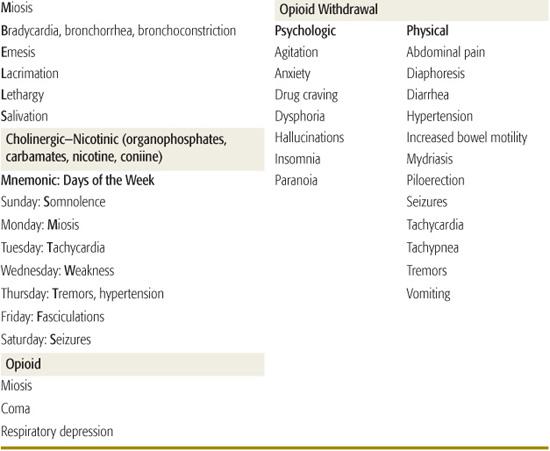
Toxicology screens should only be ordered when the result is expected to influence patient management. Urine drug-of-abuse screens detect several common illicit drugs, but are usually not necessary in the initial management of the intoxicated patient. The history and clinical toxidrome are usually adequate to narrow the differential diagnosis and guide management decisions. Comprehensive blood or urine toxicology screens have a longer turnaround time and qualitatively detect only 40 to 100 specific drugs. They will not affect emergency management, but may be helpful to the admitting physicians when the patient’s diagnosis remains unclear and the clinical course is complicated.
 RADIOLOGY
RADIOLOGY
Abdominal radiographs aid in the visualization of certain substances such as bezoars, radioopaque medications, metals, and drug-filled packets. Serial abdominal radiographs are useful to assess gastrointestinal decontamination in such patients.
 RESUSCITATION AND STABILIZATION
RESUSCITATION AND STABILIZATION
Initial treatment follows the same principles applied to other life-threatening conditions. Decreased respiratory effort, airway obstruction, and severe respiratory distress are indications for endotracheal intubation and mechanical ventilation. Judicious use of an opioid antagonist may restore normal breathing, precluding the need for endotracheal intubation. Intravenous access should be obtained promptly, and cardiovascular dysfunction corrected with intravenous fluids and pharmacologic support. Appropriate monitoring includes measurement of temperature, respiratory rate, oxygen saturation, heart rate, blood pressure, and electrocardiogram.
DECONTAMINATION
The goals of decontamination are to prevent the absorption of a toxin from the gastrointestinal tract and to remove a toxic substance from the skin or eyes. Decontamination is most effective immediately after exposure and the benefit decreases with passage of time.
 OCULAR AND DERMAL DECONTAMINATION
OCULAR AND DERMAL DECONTAMINATION
Immediate and thorough irrigation with tap water or normal saline may ameliorate injury following ocular and dermal exposures. Contact lenses are removed and irrigation of the eyes performed rapidly to prevent ocular damage.
Table 120-3. Causes of High Anion Gap Metabolic Acidosis

The patient’s clothing, jewelry, and other items should be removed for dermal decontamination and care is required to protect healthcare staff from secondary toxin exposure.
 SYRUP OF IPECAC
SYRUP OF IPECAC
Syrup of ipecac is a potent emetic that is no longer recommended as a home remedy for ingestions. The efficacy of syrup of ipecac in improving clinical outcomes remains unproved, and its use may delay the use of other decontamination techniques such as activated charcoal.
 ACTIVATED CHARCOAL
ACTIVATED CHARCOAL
Activated charcoal administered into the gastrointestinal tract binds most toxins, decreasing systemic absorption. It is ineffective for iron and other metals, lithium and other small ions, cyanide, alcohols, and most caustics. The dose of activated charcoal is 1 g/kg by mouth or via nasogastric tube for children and 50 to 100 g for adolescents and adults. A potential complication of charcoal administration is pulmonary aspiration. If a nasogastric tube is necessary, consideration must be given as to whether the expected benefit exceeds the risk. Patients with protracted vomiting are not candidates for activated charcoal, and those with decreased consciousness or undergoing sedation should not receive charcoal unless the airway has been protected with an endotracheal tube.
 GASTRIC LAVAGE
GASTRIC LAVAGE
Orogastric lavage should be considered in patients who present to medical attention within one to two hours after ingestion of a potentially lethal toxin. It is not routinely recommended for all overdose patients due to the risk of complications, including visceral perforation, tracheal placement of the orogastric lavage tube, and aspiration of vomit. Contraindications include a patient with depressed mental status and an unprotected airway and ingestion of a caustic or hydrocarbon. A large bore lavage tube (pediatrics, 16–28 Fr; adults, 36–40 Fr) is placed orogastrically with the patient in the left lateral decubitus position to optimize gastric decontamination and reduce the risk of aspiration. The stomach is lavaged with 50 to 250 mL aliquots of water or saline until clear of residual toxin.
 WHOLE BOWEL IRRIGATION
WHOLE BOWEL IRRIGATION
Whole bowel irrigation reduces the absorption of ingested toxins by decreasing gastrointestinal transit time. Polyethylene glycol electrolyte solution is administered via nasogastric tube at rates of 25 to 40 mL/kg/hour for children and 1.5 to 2 L/hour for adults until rectal effluent is clear. Whole bowel irrigation should be stopped if no rectal effluent occurs after several hours. Indications include ingestion of delayed-release medications, substances not adsorbed by charcoal, or drug-filled packages in drug smugglers (“body packers”).
ENHANCED ELIMINATION
Enhanced elimination procedures hasten the excretion of toxins from the systemic circulation at a rate greater than endogenous clearance. Interventions include urinary alkalization, multiple-dose activated charcoal, hemodialysis, and hemoperfusion.
 MULTIPLE-DOSE ACTIVATED CHARCOAL
MULTIPLE-DOSE ACTIVATED CHARCOAL
Multiple-dose activated charcoal enhances elimination and decontaminates the gastrointestinal tract by: (1) disrupting enterohepatic recirculation of the toxin; (2) utilizing the intestinal mucosa as a dialysis membrane (“gut dialysis”) to draw the toxin from the bloodstream into the intestinal lumen; and (3) binding any toxin present in the gut. It enhances elimination of phenobarbital, salicylate, quinidine, theophylline, carbamazepine, and dapsone. The dose of activated charcoal without cathartic administration is 0.5 to 1 g/kg by mouth or via nasogastric tube every 4 hours. It should be discontinued if ileus develops. Complications include charcoal aspiration, formation of charcoal bezoars, and intestinal obstruction.
 URINARY ALKALINIZATION
URINARY ALKALINIZATION
Urinary alkalinization enhances elimination of drugs that are weak acids by trapping the ionized drug in the renal tubule, where it is subsequently excreted in the urine (eFig. 120.1  ). Salicylate poisoning is the most common indication for urinary alkalinization. Sodium bicarbonate is administered in a dose of 1 to 2 mEq/kg intravenously followed by a continuous infusion titrated to a urinary pH of 7.0 to 8.0. Hypokalemia must be corrected because it impedes urinary alkalinization as potassium is reabsorbed in exchange for hydrogen ions.
). Salicylate poisoning is the most common indication for urinary alkalinization. Sodium bicarbonate is administered in a dose of 1 to 2 mEq/kg intravenously followed by a continuous infusion titrated to a urinary pH of 7.0 to 8.0. Hypokalemia must be corrected because it impedes urinary alkalinization as potassium is reabsorbed in exchange for hydrogen ions.
 HEMODIALYSIS AND HEMOPERFUSION
HEMODIALYSIS AND HEMOPERFUSION
Hemodialysis can be utilized to correct fluid, electrolyte, and acid–base imbalances and to hasten the removal of some toxic substances from the blood. It is effective for toxins with small volumes of distribution (< 1.0 L/kg), low molecular weight, and low protein binding (Table 120-4). Additional indications include a blood level of the toxin associated with severe toxicity or death, impairment of the natural mechanism of elimination, deteriorating clinical condition despite maximal supportive care, and toxins with severe delayed effects.
Table 120-4. Agents Removed by Hemodialysis
Mnemonic: I STUMBLE |
Isopropanol |
Salicylates |
Theophylline |
Uremia |
Methanol |
Barbiturates |
Lithium |
Ethylene glycol |
Hemoperfusion is similar to hemodialysis except for substitution of the dialysis membrane by a cartridge containing an adsorbent material such as charcoal. Hemoperfusion often achieves greater clearance rates than hemodialysis and is more effective for larger molecules because there is no ultrafiltration.
 ANTIDOTES
ANTIDOTES
An antidote is an agent that prevents or reverses the effects of poisoning. Most are dispositional antidotes that decrease toxicity by altering absorption, distribution, metabolism, or elimination. Competitive antagonists interfere with the toxin binding to receptor sites. Physiologic antagonists alter the physiologic effect of the toxin. Antidotes are available for a very limited number of poisonings and do not replace meticulous supportive care and good clinical judgment. The risks and benefits of each antidote must be carefully considered.
DISPOSITION
Asymptomatic patients with a potential toxic exposure are observed 4 to 8 hours from the time of exposure. If symptoms develop, the patient is admitted to the hospital for further management. Any patient with severe toxicity will require admission to an intensive care unit. A longer observation period is required for toxins with delayed onset of action such as sustained-release products, agents that are converted into toxic metabolites, and drugs with delayed distribution into the tissue compartment. Cellular injury may begin early with delayed onset of overt symptoms, such as with acetaminophen.
All poisonings in children require social evaluation to determine home safety and provide poison prevention education. Patients may have been intentionally poisoned as a form of physical abuse or for purposes of sexual exploitation. Any intentional overdose warrants psychiatric evaluation to evaluate suicidal intent. Patients with chemical dependency are referred to an appropriate treatment program.
PHARMACEUTICAL AGENTS
ACETAMINOPHEN
Acetaminophen (N-acetyl-p-aminophenol or APAP), a widely used analgesic and antipyretic agent, is responsible for 10% of all calls to poison control centers. After therapeutic dosing, acetaminophen undergoes glucuronidation and sulfation in the liver prior to urinary and bile excretion (see Fig. 120-1). A small percentage is oxidized in the hepatocytes by the cyto-chrome P450 system of CYP oxidases to produce N-acetyl-p-benzoquinoneimine (NAPQI), a highly reactive two-electron species that can act as an oxidant and thus is highly cytotoxic. NAPQI is eliminated by conjugation to the reduced form of glutathione (GSH), and the resulting APAP-GSH conjugate is eliminated as mercapturic and cysteine conjugates. After an overdose, however, the amount of NAPQI generated can exceed the capacity of this detoxification mechanism. NAPQI then accumulates in the hepatocytes and toxicity ensues (Fig. 120-1). Acute ingestion of more than 150 to 200 mg/kg in children or 7.5 g in adults is potentially hepatotoxic. Chronic or subacute overdose also results in liver injury.
The clinical presentation of acute acetaminophen toxicity is described in 4 phases. Phase I occurs during the first few hours after ingestion, and is characterized by gastrointestinal symptoms of nausea, vomiting, and abdominal pain, but some patients are asymptomatic. Phase II occurs within 24 hours as the acute gastrointestinal symptoms subside. The onset of hepatotoxicity is manifested by upper-right quadrant abdominal tenderness and elevation of laboratory markers of liver injury such as transaminases and prothrombin time (PT). Phase III is the period of maximal hepatotoxicity, usually occurring within 72 to 96 hours. In mild cases, spontaneous recovery occurs. In severe cases, fulminant hepatic failure is manifested by coagulopathy, encephalopathy, and coma. Fatalities typically occur between 3 and 5 days because of multiorgan failure, hemorrhage, respiratory failure, sepsis, and cerebral edema. Phase IV is the period of hepatic recovery, which may take a week or longer, depending on the severity of the liver injury.
Treatment decisions are guided by plotting the serum acetaminophen concentration on the Rumack-Matthew nomogram (Fig. 120-2) at 4 hours or later after an acute ingestion. In the United States, the usual recommendation is to initiate antidotal therapy if the acetaminophen level is above the lower (possible hepatotoxicity) line on the nomogram because this is the safer course of action. Antidotal therapy with N-acetylcystine (NAC) should be started within 8 hours of drug ingestion because its efficacy for preventing fulminant hepatic failure diminishes thereafter.
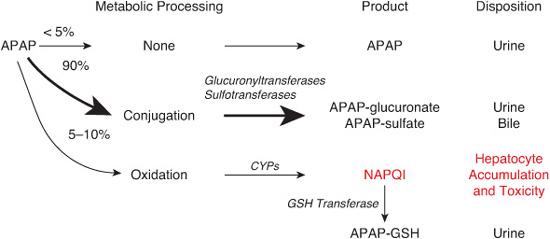
FIGURE 120-1. Mechanism of acetaminophen (N-acetyl-p-aminophenol or APAP) toxicity. See text for details.
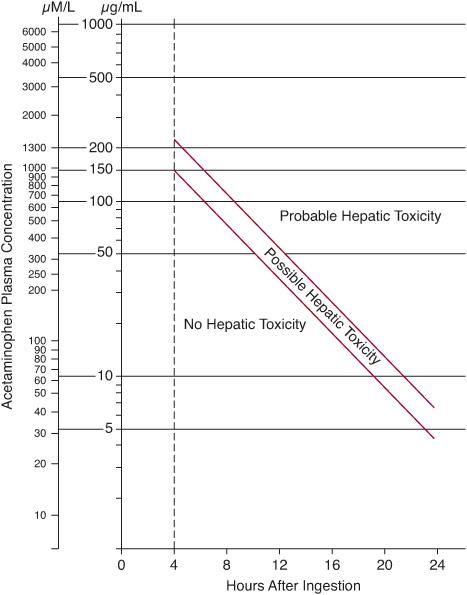
FIGURE 120-2. Rumack-Matthews nomogram showing the risk of hepatotoxicity based on the relationship between plasma acetaminophen concentration and time after ingestion. The range between the red lines represents a 25% chance of clinically significant disease.
NAC functions both as a glutathione precursor to replenish depleted glutathione stores and as a glutathione substitute that directly binds to NAPQI. In addition, NAC facilitates the sulfation pathway of acetaminophen metabolism. The oral NAC protocol consists of a loading dose of 140 mg/kg followed by 17 subsequent doses of 70 mg/kg administered every 4 hours over a 72-hour period. Treatment may be discontinued at 24 hours if there has been no elevation of liver transaminases and acetaminophen is no longer detectable in the serum. There are several intravenous N-acetylcysteine regimens that have been utilized, including 21-, 36-, and 48-hour protocols. The commonly used 21-hour protocol consists of a loading dose of 150 mg/kg administered over 1 hour, followed by an additional 50 mg/kg over the next 4 hours, and a subsequent 100 mg/kg over the next 16 hours.1
Laboratory monitoring includes measuring the serum acetaminophen level, liver transaminases, electrolytes, blood urea nitrogen, creatinine, and prothrombin time. Patients with severe hepatotoxicity require early evaluation for liver transplantation.
ANTICOAGULANTS
Warfarin (Coumadin) was initially used as a rodenticide before its therapeutic applications were recognized. Rats and mice quickly became resistant, prompting the development of more potent second-generation superwarfarins. Rodenticides are typically marketed in very low concentration pellets, requiring multiple exposures to exterminate the rodent. These compounds inhibit vitamin K, a cofactor required for the synthesis of coagulation factors II, VII, IX, and X.
The typical clinical scenario for warfarin ingestion would be a toddler who has ingested a small amount of rodenticide. In this situation, no significant anticoagulation is expected, and no interventions are required.2 In contrast, larger ingestions with suicidal intent or chronic exposures may result in poisoning. The anticoagulant effect will not be seen until the circulating clotting factors have been degraded, which may take 1 to 2 days. Excessive anticoagulation results in ecchymoses, bleeding gums, subconjunctival hemorrhage, hematemesis, melena, or hematuria. In severe cases, intracranial hemorrhage or gastrointestinal bleeding is life threatening. The duration of effect after a single dose of warfarin is 2 to 7 days, whereas coagulopathy from superwarfarin poisoning often persists for months due to an extremely long elimination half-life.
Laboratory monitoring includes evaluation of the prothrombin time (PT) and International Normalized Ratio (INR). A normal PT 48 hours after ingestion rules out poisoning. With more significant exposures, a complete blood count, blood type, and crossmatch are obtained. Severe hemorrhage is treated with fresh-frozen plasma, packed red blood cells, and other blood products as needed. Administration of vitamin K1 (phytonadione) allows regeneration of clotting factors, but peak effect may take 24 hours or longer. Prolonged coagulopathy may require repeated administration of vitamin K1 and fresh-frozen plasma. Vitamin K1 is recommended for documented coagulopathy, but should not be administered prophylactically because it will mask the development of toxicity and make the end point of treatment difficult to assess.
ANTICONVULSANTS
The anticonvulsants are a diverse group of medications with different mechanisms of action and toxicities in overdose. They are used for seizure control and other conditions such as mood disorders and refractory pain syndromes. Carbamazepine, phenobarbital, phenytoin, and valproic acid are most commonly responsible for acute intoxications.
 CARBAMAZEPINE
CARBAMAZEPINE
Carbamazepine’s primary therapeutic mechanism of action is sodium channel inhibition in the central nervous system. Toxic manifestations after an overdose are similar to those produced by tricyclic antidepressants, with which carbamazepine has structural analogies, resulting in central nervous system depression, anti-cholinergic effects, and myocardial sodium channel inhibition. Induction of vasopressin secretion may result in the syndrome of inappropriate secretion of antidiuretic hormone.
Elevated serum carbamazepine levels are associated with neurologic abnormalities including nystagmus, ataxia, dysarthria, myoclonus, and dystonia. Levels above 40 mg/dL are associated with severe toxicity, including coma, respiratory depression, and seizures. Cardiovascular disturbances include tachycardia, hypotension, myocardial depression, and cardiac conduction abnormalities such as prolongation of the QRS complex and QT interval. Mydriasis, hyper-thermia, ileus, and other signs of cholinergic inhibition may occur.
Monitoring after an ingestion should include obtaining serum carbamazepine level every 4 to 6 hours until a downward trend is established. Serum electrolytes should be checked for hyponatremia and electrocardiogram monitoring helps detect cardiotoxicity. Treatment is supportive. Enhanced elimination procedures may be considered in patients with severe toxicity that is not responsive to standard supportive care.
 PHENOBARBITAL
PHENOBARBITAL
Barbiturates depress central nervous system function by enhancing the neuronal inhibition mediated by gamma-aminobutyric acid (GABA). Supratherapeutic serum phenobarbital levels are associated with progressive ataxia, dysarthria, nystagmus, sedation, coma, hypothermia, and respiratory depression. Decreased sympathetic tone results in bradycardia and hypotension. A serum phenobarbital level should be obtained serially until it is documented to be decreasing. Treatment is supportive. Enhanced elimination may benefit patients with severe toxicity refractory to supportive care.
 PHENYTOIN
PHENYTOIN
Phenytoin and fosphenytoin are nonsedating at therapeutic doses. Phenytoin decreases the activity of neuronal voltage-dependent sodium channels, resulting in suppression of seizure activity. Excessive doses result in ataxia, nystagmus, diplopia, dysarthria, confusion, coma, and respiratory depression. Cardiotoxicity has not been reported following oral overdoses of phenytoin, although rapid intravenous administration may cause bradycardia and hypotension.
Phenytoin is poorly water soluble, and the intravenous preparation requires a solution of 40% propylene glycol and 10% ethanol at a pH of 12. Rapid infusion (> 50 mg/min phenytoin) results in myocardial depression and decreased peripheral vascular resistance largely owing to propylene glycol. Local irritation and tissue damage may be caused by the propylene glycol and the alkaline pH of the intravenous formulation. Fosphenytoin is a water-soluble product that is converted into phenytoin following intravenous administration. It is buffered to a pH of 8 to 9 and does not contain propylene glycol, allowing rapid intravenous administration (up to 150 mg/min of phenytoin equivalents). However, excessive doses of fosphenytoin have been reported to cause myocardial depression.
A serum phenytoin level should be obtained serially until a downward trend is documented. Management is supportive. Multiple-dose activated charcoal may enhance the elimination of phenytoin, but is usually not necessary because of the success of supportive care alone.
 VALPROIC ACID
VALPROIC ACID
Valproic acid inhibits voltage-gated sodium channels and increases GABA levels in the central nervous system, resulting in central nervous system depression. In addition, valproic acid interferes with fatty acid metabolism by causing carnitine deficiency, impaired mitochondrial beta-oxidation, and disruption of the urea cycle.
Patients with valproic acid toxicity present with confusion and lethargy, which may progress to coma, cerebral edema, and respiratory failure. Severe toxicity includes hypotension, metabolic acidosis, hypernatremia, hypocalcemia, pancreatitis, hepatic injury, hyperammonemia, renal insufficiency, and bone marrow suppression. Elevated ammonia levels result in valproate-induced hyperammonemic encephalopathy, characterized by altered mental status, focal neurologic abnormalities, and seizures.3
A serum valproic acid level is obtained serially until a downward trend is established. Other useful laboratory studies include blood gas analysis, complete blood count, serum electrolytes, glucose, blood urea nitrogen, creatinine, calcium, liver transaminases, bilirubin, prothrombin time, lipase, amylase, lactate, ammonia, and carnitine. Treatment with carnitine is recommended if hyperammonemia, hepatotoxicity, or carnitine deficiency is present. Enhanced elimination techniques may be considered in patients with severe toxicity.
 ANTIPSYCHOTICS
ANTIPSYCHOTICS
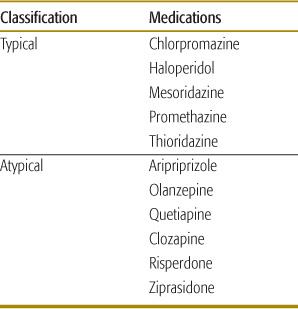
The antipsychotic medications are classified as typical or atypical (Table 120-5). The typical antipsychotics were introduced in the 1950s. Soon they were associated with serious adverse effects including extrapyramidal syndromes (particularly dyskinesia in children), tardive dyskinesia, and neuroleptic malignant syndrome. Noncompliance and treatment failures were common. Some of these medications are nowadays prescribed because of their non-psychoactive properties, such as the antiemetic effect of promethazine.
The atypical antipsychotics were developed to overcome the shortcomings of the traditional antipsychotic medications, and were first marketed in the 1980s. They are less likely to cause extrapyramidal syndromes or tardive dyskinesia and they are less toxic following acute overdose.
The therapeutic mechanism of typical antipsychotic agents is antagonism of D2-dopaminergic receptors in the mesolimbic system. However, nonselective antagonism of D2 receptors in other dopaminergic pathways may result in extrapyramidal side effects, including acute dystonia, akathisia, parkinsonism, neuroleptic malignant syndrome, and tardive dyskinesia. Adverse cardiovascular effects include alpha-adrenergic receptor antagonism, blockade of myocardial sodium channels, and inhibition of the delayed rectifier current. Typical antipsychotics can have anticholinergic activity and cause lowered seizure threshold and impaired temperature regulation.
Table 120-5. Classification of Selected Antipsychotic Agents
Atypical antipsychotics are more selective antagonists of mesolimbic D2 receptors with a lower binding potency, resulting in fewer extrapyramidal side effects. Therapeutic benefit also results from blockade of serotonergic 5-HT1A and 5-HT2A receptors. Adverse effects are due to inhibition of α-adrenergic, H1 histamine, and M1 muscarinic receptors.4
The clinical presentation of mild intoxication includes confusion, sedation, vagolysis, and orthostatic hypotension with reflex tachycardia. Severe overdoses result in coma, seizures, respiratory arrest, and hypothermia or hyperthermia. Hypotension occurs secondary to impaired myocardial contractility and peripheral vasodilation. Conduction abnormalities owing to impaired sodium and potassium channel activity include widened QRS and QTc interval prolongation. Ventricular tachycardia including torsade de pointes may result.
Extrapyramidal dystonic reactions include tremor, rigidity, bradykinesia, and spasms of the muscles of the eyes (oculogyric crisis), face, tongue, lips, jaw, neck (torticollis), abdomen (tortipelvis), and spine (opisthotonus). Neuroleptic malignant syndrome (NMS) is a life-threatening condition characterized by altered mental status, muscular rigidity, hyperthermia, and autonomic dysfunction.
Diagnosis is based on the clinical presentation. Quantitative blood levels are not routinely available. Comprehensive urine toxicology screens may detect only phenothiazines. Continuous electrocardiogram monitoring is essential to detect cardiac conduction abnormalities and dysrhythmias.
Management is primarily supportive. Specific interventions include sodium bicarbonate for treatment of myocardial sodium channel blockade and QRS interval prolongation and magnesium infusion or overdrive pacing for treatment of torsade de pointes. Dystonic reactions are treated with diphenhydramine or benztropine. Management of neuroleptic malignant syndrome requires aggressive cooling measures, intravenous hydration, advanced airway management as indicated, and intravenous benzodiazepines for sedation and muscle relaxation. Dantrolene (a muscle relaxant) and bromocriptine (a dopamine agonist) may be of benefit in the treatment of neuromalignant syndrome.
BETA-BLOCKERS AND CALCIUM CHANNEL BLOCKERS
Beta-adrenergic blocker (BB) and calcium channel blocker (CCB) medications are discussed together because of similarities in clinical presentation and management after overdose. Many of these similarities are owing to their common effect of inhibiting the influx of calcium into myocardial cells, where it participates in physiologic signaling and other critical processes. Severe poisoning is difficult to treat and is associated with significant morbidity and mortality.
Beta-blockers inhibit the binding of catecholamines to β-adrenergic receptors. Normally, myocardial β1-adrenergic receptor stimulation results in activation of adenyl cyclase, which catalyzes the formation of cyclic adenosine monophosphate (cAMP), which in turn promotes phosphorylation and opening of calcium channels. Blockade of cardiac β1-adrenergic receptors results in decreased chronotropy and inotropy. Blockade of noncardiac β2-adrenergic receptors antagonizes bronchial and smooth muscle relaxation, and also causes hypoglycemia by inhibition of glycogenolysis and gluconeogenesis.
Calcium channel blockers bind to and inhibit the influx of calcium through the L-type calcium channels of myocardial cells, resulting in decreased chronotropy and inotropy. Antagonism of similar vascular smooth muscle calcium channels results in coronary and peripheral vasodilation. Calcium-mediated insulin release from pancreatic β-islet cells is inhibited. This prevents myocardial cells from utilizing glucose as an energy source, resulting in further myocardial dysfunction. The resulting shock state resembles diabetic ketoacidosis, with hyperglycemia and acidemia.
The hallmark of beta-blocker and calcium channel blocker poisoning is cardiovascular depression manifested by bradycardia, heart block, impaired cardiac contractility, hypotension, and profound shock. A widened QRS duration may be caused by beta-blockers such as propranolol that also inhibit sodium channels. Altered mental status is usually caused by cerebral hypoperfusion, but in the case of beta-blockers may also be caused by hypoglycemia. Beta-blockers such as propranolol that cross the blood-brain barrier and have membrane-depressant effects may cause coma, convulsions, and respiratory arrest. Noncardiac effects of calcium channel blockers include nausea, vomiting, and hyperglycemia. Global hypoperfusion may result in angina or myocardial infarction, cerebral ischemia, hepatic dysfunction, renal failure, and metabolic acidosis.
Diagnosis of beta-blocker and calcium channel blocker poisoning is based on the history and clinical presentation, especially bradycardia and hypotension. Beta-blocker poisoning may cause hypoglycemia, whereas calcium channel blocker poisoning typically results in hyperglycemia. Studies that are helpful in assessment and management include electrolytes, glucose, blood urea nitrogen, creatinine, blood gas analysis, and electrocardiogram monitoring. A rapid bedside cardiac ultrasound is useful to assess myocardial function. Patients with severe poisoning may require invasive monitoring to guide management.
Gastric lavage and administration of activated charcoal may be helpful early in patients with large recent ingestions. If the patient already has significant bradycardia, hypotension, or altered mental status, resuscitative procedures take precedence. Whole bowel irrigation and multiple-dose activated charcoal should be considered in patients with ingestion of sustained-release preparations. General supportive measures include atropine for bradycardia and intravenous fluids for hypotension. Calcium and catecholamines should be used initially but may be ineffective after calcium channels and β-adrenergic receptors are inhibited. Glucagon is a specific antidote for beta-blocker poisoning and has been used with some success in calcium channel blocker overdose as well. Glucagon produces positive inotropic and chronotropic effects by stimulating adenyl cyclase and opening calcium channels independent of the β-adrenergic receptor activation. Amrinone increases intracellular cAMP and calcium influx to improve myocardial contractility, but may exacerbate hypotension by causing peripheral vasodilation. Vasopressin improves blood pressure and increases the response to catecholamines. Hyperinsulinemia/euglycemia therapy improves myocardial contractility in calcium channel blocker overdose, often when other pharmacologic therapy has failed. It is postulated that this therapy corrects the impaired myocardial substrate utilization caused by calcium channel blocker–induced hypoinsulinemia, resulting in improved carbohydrate metabolism and increased myocardial contractility.5 Other extraordinary measures have been used to maintain perfusion while waiting for cardiac drug toxicity to abate. When pharmacologic therapy has failed, cardiopulmonary bypass or extracorporeal membrane oxygenation may be life-saving.
CLONIDINE
Clonidine is a centrally acting antihypertensive agent that is also prescribed for conditions such as attention deficit hyperactivity disorder and drug withdrawal. Clonidine binds to inhibitory presynaptic α2-adrenergic receptors in the medulla, resulting in decreased sympathetic outflow from the central nervous system.
The hallmark of intoxication is generalized sympathetic depression. Neurologic manifestations are similar to the opioid toxidrome, including pinpoint pupils, hypotonia, sedation, coma, hypothermia, respiratory depression, and apnea. Cardiovascular toxicity is manifested by bradycardia, decreased cardiac output, and hypotension.
Although clonidine poisoning mimics the opioid toxidrome, it does not respond to naloxone. Sinus bradycardia does not require treatment unless it is associated with hypotension and poor perfusion. Atropine will often improve heart rate and cardiac output. Persistent hypotension is treated with intravenous fluids and catecholamine infusion. Coma and respiratory depression may require advanced airway management. Gastrointestinal decontamination is usually not indicated because of rapid onset of symptoms and good outcome with supportive care.
CYCLIC ANTIDEPRESSANTS
Tricyclic antidepressants (TCAs) were first drugs used to treat depression in the 1950s, but tricyclic antidepressant overdoses caused severe toxicity, resulting in many poisoning deaths. The selective serotonin reuptake inhibitors (SSRIs) were introduced in the 1980s and have a better safety profile than the tricyclic antidepressants.
The therapeutic mechanism of tricyclic anti-depressants is inhibition of reuptake of biogenic amine neurotransmitters in the central nervous system, including norepinephrine, dopamine, and serotonin. In overdose, other mechanisms result in toxicity, including antagonism of muscarinic cholinergic receptors, α1-adrenergic receptors, serotonin receptors, histaminic H1 and H2 receptors, GABAA receptors, and myocardial fast sodium channels.
The therapeutic mechanism of serotonin reuptake inhibitors is serotonin reuptake inhibition. These medications do not usually cause severe toxicity except in large overdose. However, excessive serotonergic activity may result in the life-threatening serotonin syndrome (see later in this section). Some serotonin reuptake inhibitors cause additional toxicity via inhibition of dopamine and norepinephrine reuptake and antagonism of α1-adrenergic receptors. Some agents also have sodium and potassium channel inhibition causing QRS and QTc prolongation.
Patients with significant tricyclic antidepressants ingestions usually manifest symptoms within 30 to 60 minutes, and life-threatening toxicity within 6 hours. The most common cause of death is cardiovascular toxicity. Inhibition of myocardial fast sodium channels causes slowed phase 0 depolarization, decreased contractility, widened QRS and QT intervals, and ventricular tachycardia including torsades de pointes. Severe hypotension is caused by norepinephrine depletion, α1-adrenergic receptor blockade, myocardial depression, and dysrhythmias. Muscarinic cholinergic receptor blockade results in sedation, delirium, coma, myoclonus, and seizures. Other anticholinergic effects include tachycardia, hyperthermia, mydriasis, ileus, urinary retention, diminished sweating, and dry mucus membranes. Seizures are also caused by inhibition of GABAA receptors.
Significant SSRI overdose results in nausea, ataxia, and sedation. Coma and respiratory depression may occur, particularly when alcohol or other sedating drugs are also involved. Some agents, such as buproprion, may cause tremor, agitation, and seizures. Cardiovascular effects include sinus tachycardia, hypotension, and cardiac conduction disturbances, but are usually not life threatening.
The most common severe toxicity associated with SSRIs is the serotonin syndrome, caused by excessive stimulation of serotonin 5-HT2A and possibly 5-HT1A receptors. The serotonin syndrome often occurs when an SSRI is combined with another drug with serotonergic activity, but it has been reported in patients taking single or multiple SSRIs. Its manifestations include agitation, altered mental status, coma, incoordination, myoclonus, hyperreflexia, tremor, muscle rigidity, diarrhea, diaphoresis, and hyperthermia. Patients may only exhibit a few of the symptoms, making the diagnosis challenging. Deaths are caused by hyperthermia, rhabdomyolysis, lactic acidosis, disseminated intravascular coagulation, and multiorgan failure.
The diagnosis of cyclic antidepressant poisoning is based on the history and clinical presentation. In the patient with an unknown ingestion, the combination of anticholinergic signs, coma, seizures, hypotension, and a widened QRS interval strongly suggests tricyclic antidepressant poisoning.
Because of the potential lethality of tricyclic antidepressant poisoning, gastrointestinal decontamination procedures such as orogastric lavage and activated charcoal are considered for patients who present early after an overdose. Patients presenting later with severe symptoms are unlikely to benefit from gastrointestinal decontamination.
The management of cyclic antidepressant poisoning is mainly supportive. Intravenous access and cardiac monitoring should be immediately established. Endotracheal intubation is indicated if significant central nervous system depression is present. Hypotension should be treated with rapid crystalloid infusion and pharmacologic support. Norepinephrine is recommended because of its predominant α1-mediated vasoconstrictive effects.
Seizures are treated aggressively because the resultant hypoxia and acidosis potentiate tri-cyclic antidepressant-induced cardiotoxicity. Anticonvulsants that enhance gamma-aminobutyric acid such as benzodiazepines or barbiturates are usually effective.
It is important to monitor the electrocardiogram and QRS interval. A QRS duration of greater than 100 msec is associated with seizures and a duration of greater than 160 msec is associated with ventricular dysrhythmias. Ventricular dysrhythmias and hypotension are treated with serum alkalinization and sodium loading.6 Increasing the extracellular sodium concentration helps overcome the sodium channel blockade caused by tricyclic antidepressants, and serum alkalinization diminishes drug binding to the fast sodium channel. A QRS interval greater than 100 msec, ventricular dysrhythmias, or intractable hypotension should be treated with intravenous sodium bicarbonate in a dose of 1 to 2 mEq/kg, repeated as necessary to maintain a serum pH between 7.45 and 7.55. Use of hypertonic saline (3% NaCl) to increase the extracellular sodium concentration or hyperventilation to induce a respiratory alkalosis may also be considered in difficult cases. Ventricular dysrhythmias that fail to respond to sodium bicarbonate may be treated with lidocaine, a class IB antidysrhythmic agent. Although lidocaine theoretically can worsen ventricular dysrhythmias, it has been used safely in tricyclic antidepressants poisoning. However, phenytoin (another class IB antidysrhythmic agent) is not recommended because it has not been well studied and may worsen ventricular dysrhythmias. Class IA and IC antidysrhythmic agents are contraindicated because they exacerbate tricyclic antidepressant–induced cardiotoxicity.
Patients with cardiogenic shock who fail to respond to maximal therapeutic interventions are candidates for extracorporeal life support such as cardiopulmonary bypass or extracorporeal membrane oxygenation. Case reports indicate that these measures favor survival as the tricyclic antidepressant is eliminated from the body and the cardiotoxicity abates.
Serotonin syndrome is diagnosed when consistent symptoms occur in the setting of serotonergic medication use and other etiologies are excluded. Treatment consists of immediate withdrawal of any offending drugs and supportive care that focuses on decreasing hyperthermia and muscular rigidity. Effective measures include external cooling, intravenous hydration, and benzodiazepines or neuromuscular blockade to achieve muscle relaxation. Case reports support cyproheptadine to treat serotonin syndrome owing to its inhibitory effects at 5-HT1A and 5-HT2A serotonin receptors.
DIGOXIN AND CARDIAC GLYCOSIDES
Digoxin and related cardiac glycosides are used to treat congestive heart failure and control ventricular rate in supraventricular tachydysrhythmias. The narrow therapeutic index of digoxin increases the risk of toxicity during therapeutic administration and with medication errors. Other sources of exposure include unintentional and intentional overdose, as well as ingestion of plants and other natural sources of cardiac glycosides.
Cardiac glycosides inhibit the sodium-potassium-adenosine-triphosphatase (Na+/K+-ATPase) pump. This mechanism is used to treat congestive heart failure because it increases the intracellular calcium in myocardial cells, resulting in improved contractility. During normal repolarization, the Na+/K+-ATPase restores the resting membrane potential by pumping potassium ions into the myocardial cell in exchange for sodium ions out of the cell. When this pump is inhibited by cardiac glycosides, the result is an increase in intracellular sodium and an increase in extracellular potassium. The increased intra-cellular sodium inhibits efflux of calcium through the Na+/Ca2+ antiporter at the cell membrane. The result is an increase in intracellular calcium that results in improved inotropy.
In addition, digoxin enhances vagal tone, decreasing the rate of depolarization and conduction through the sinoatrial (SA) and atrioventricular (AV) nodes, and increasing the refractory period. During digoxin poisoning, excessive increases in intracellular calcium results in oscillatory disturbances of membrane potential, resulting in ectopy and tachydysrhythmias.
The clinical presentation of acute digoxin poisoning includes cardiac and noncardiac features. The noncardiac effects include gastrointestinal manifestations such as nausea, vomiting, and diarrhea; neurologic manifestations such as confusion, headaches, weakness, and seizures; and visual manifestations such as blurred vision, scotoma, and xanthopsia.
The cardiac effects of digoxin poisoning are the primary life-threatening concern. Bradycardia is an early manifestation. Reduced conduction velocity through the atrial conducting tissue and atrioventricular node causes a prolonged PR interval and varying degrees of atrioventricular block. This is often followed by increased myocardial automaticity and excitability, resulting in atrial, junctional, and ventricular ectopy, and tachydysrhythmias. Death occurs due to intractable dysrhythmias, hypotension, and ventricular fibrillation.
Diagnosis is based on the history of exposure and the characteristic cardiac and noncardiac effects of digoxin poisoning. The combination of bradycardia, increased automaticity, and hyper-kalemia is characteristic.
The serum digoxin level must be interpreted with caution. The correlation between clinical effects and serum digoxin level is based on the steady state concentration. It is therefore more helpful in predicting toxicity in the patient with chronic overdose. Although the therapeutic range for serum digoxin is usually between 0.5 and 2 ng/mL, up to 10% of patients in this range may demonstrate toxicity. Digoxin demonstrates a biphasic distribution pattern following acute overdose. Serum levels will initially be elevated without clinical toxicity because distribution into the tissues occurs over many hours. Digoxin levels 10 times greater than the therapeutic serum concentration can be seen in patients during the first few hours after acute overdose without apparent toxicity. Similar concentrations in a patient with chronic toxicity would be fatal. Serum digoxin concentrations then decline as the drug is distributed into the tissues and eliminated by the kidneys, and it is during this phase that toxicity may begin.
Management of digoxin poisoning begins with standard supportive care and continuous electrocardiogram monitoring. Intravenous access is obtained, and blood analyzed for serum digoxin, electrolyte, creatinine, calcium, and magnesium concentrations.
The treatment of choice for serious digoxin (or other cardioactive steroids) poisoning is intravenous administration of digoxin-specific Fab antibody fragments.7 Indications include life-threatening dysrhythmia, conduction delay, hyperkalemia (> 5 mEq/L), cardiogenic shock, or cardiac arrest due to digoxin overdose. Other indications include a high likelihood of progression to life-threatening digoxin toxicity, such as ingestion of more than 4 mg or 0.1 mg/kg in a child, ingestion of more than 10 mg in an adult, serum digoxin level greater than or equal to 15 ng/mL at any time or greater than or equal to 10 ng/mL 6 hours after ingestion, or rapidly progressive signs and symptoms of digoxin poisoning.
Formulas are available to calculate the dose of digoxin-specific Fab based on the serum digoxin concentration or the known amount of digoxin that was ingested. However, the history of the amount ingested is notoriously inaccurate and after an acute exposure, the serum digoxin concentration does not reflect steady-state distribution. It is more practical to administer an empiric dose of 10 to 20 vials of digoxin-specific Fab for acute overdose, repeating as needed to reverse life-threatening toxicity. A lower dose is recommended for chronic digoxin poisoning to reduce the toxic effects by neutralizing a fraction of the digoxin while maintaining therapeutic benefits in the patient with underlying cardiac disease. Empiric dosing of digoxin-specific Fab for chronic poisoning is 1 to 2 vials for children and 3 to 6 vials for adults, titrated to clinical effect.
Following administration of digoxin-specific Fab, total serum digoxin levels increase dramatically because digoxin is pulled from the tissue compartment into the intravascular space, where it is bound by the Fab fragments and inactivated. The complex is subsequently excreted by the kidneys. Most laboratory assays measure total digoxin including the digoxin-Fab complex, which is not generally useful after administration of digoxin-specific Fab. However, measurement of free serum digoxin levels may be helpful in guiding further therapy.
A crucial aspect in the treatment of digoxin poisoning is the management of potassium, magnesium, and calcium homeostasis. Hyper-kalemia is a consequence of digoxin poisoning and may be treated with sodium bicarbonate or glucose and insulin to shift potassium into the cells. Administration of calcium is contraindicated because digoxin has already caused an excessive influx of calcium into myocardial cells. Administration of additional calcium increases the incidence of dysrhythmias. Although digoxin initially causes an elevated extracellular potassium concentration, increased renal elimination leads to depleted total body stores. Serum potassium concentrations often fall precipitously following digoxin-specific Fab and must be checked frequently.
Hypokalemia is especially dangerous in digoxin poisoning because it exacerbates cardiotoxicity. A fall in serum potassium concentration from 3.5 mEq/L to 3 mEq/L increases cardiac sensitivity to digoxin by approximately 50%. Patients with chronic digoxin toxicity are often hypokalemic because of concomitant treatment with diuretics. Significant hypokalemia should be corrected cautiously, especially in the patient with renal insufficiency.
Magnesium plays an important role in digoxin toxicity because it is a cofactor for the Na+/K+-ATPase pump. Hypomagnesemia potentiates digoxin-induced cardiotoxicity and inhibits correction of hypokalemia. Patients who are on chronic digoxin therapy may be hypomagnesemic because of concurrent diuretic use. Intracellular magnesium depletion may be present despite a normal serum magnesium concentration. Magnesium replacement is indicated for patients with digoxin-related cardiotoxicity, hypokalemia, and documented hypomagnesemia. Caution is again advised in the patient with renal insufficiency.
Gastrointestinal decontamination will not benefit the patient with chronic digoxin toxicity, but may be considered for recent acute ingestions. Activated charcoal binds to digoxin, decreasing gastrointestinal absorption. Digoxin poisoning results in conduction delays and increased vagal tone, and therefore induced emesis and gastric lavage are avoided because vagal stimulation may induce bradydysrhythmias.
IRON
Iron is found in many commonly available preparations (eTable 120.1  ) and remains an important cause of severe childhood poisoning. Its toxic effects are mediated primarily by free-radical production, resulting in multiorgan injury.
) and remains an important cause of severe childhood poisoning. Its toxic effects are mediated primarily by free-radical production, resulting in multiorgan injury.
The first stage of iron poisoning results from direct gastrointestinal toxicity and is characterized by nausea, vomiting, diarrhea, and abdominal pain. Hematemesis and hematochezia may also occur. The second stage is the latent stage, which usually occurs 6 to 24 hours after ingestion following resolution of gastrointestinal symptoms and preceding the onset of systemic toxicity. Patients may have persistent lethargy, tachycardia, and metabolic acidosis. The third stage is shock and systemic toxicity, which can occur very rapidly after a massive ingestion or up to 24 hours after a moderate ingestion. Hypoperfusion and metabolic acidosis result from decreased myocardial contractility, vasodilation, and hypovolemia secondary to gastrointestinal bleeding and fluid shifts from the vascular to extravascular space. Central nervous system involvement includes lethargy, seizures, or coma. The fourth stage is hepatic failure, which can occur 2 to 3 days after ingestion. This stage is characterized by coagulopathy, hypoglycemia, elevated transaminase levels, hyperbilirubinemia, hyperammonemia, coma, and renal failure. The fifth stage is the late complications resulting from the initial corrosive gastrointestinal injury, such as gastric outlet obstruction and small intestinal strictures. These may manifest 2 to 8 weeks following ingestion.
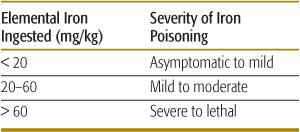
Diagnosis is based on a history of iron ingestion and the presence of consistent signs and symptoms, especially gastrointestinal distress or shock. The absence of symptoms within the first 6 hours excludes serious iron toxicity. The amount of elemental iron ingested correlates with toxicity (Table 120-6). A peak serum iron concentration obtained 4 hours after ingestion predicts the severity of toxicity. Peak iron concentrations less than 300 μg/dL are usually nontoxic, whereas levels of 300 to 500 μg/dL often result in mild to moderate toxicity. Peak iron concentrations greater than 500 μg/dL are associated with severe toxicity, and those greater than 1000 μg/dL may be lethal.
Other helpful laboratory studies include a complete blood count, blood gas analysis, electrolytes, glucose, blood urea nitrogen, creatinine, liver function tests, lactate and coagulation studies to evaluate for anemia, metabolic acidosis, liver failure, renal insufficiency, and coagulopathy. Abdominal radiographs may identify iron tablets, concretions, or free air in the abdomen.
Treatment of the patient with serious iron overdose starts with stabilization and supportive measures. Intravenous chelation with defer-oxamine should be considered for any patient with a serum iron level greater than 500 μg/dL or signs of significant toxicity such as metabolic acidosis and hypoperfusion. Deferoxamine binds iron with high affinity, forming ferrioxamine (deferoxamine-iron) that is eliminated by the kidneys. The classic vin rosé coloration of the urine does not always occur even in the presence of significant iron toxicity. Therapy may be discontinued when systemic toxicity resolves and serum iron levels return to normal. Rapid infusion of deferoxamine may result in hypotension, and prolonged deferoxamine therapy has been associated with lung injury and Yersinia enterocolitica sepsis.
Whole bowel irrigation is recommended when iron tablets are seen on radiographs of the gastrointestinal tract. Serial abdominal radiographs are obtained to document elimination of the iron tablets. Gastric lavage is ineffective because iron tablets are too large and adherent to be removed. Activated charcoal does not bind iron. Surgery may be required to excise adherent iron concretions that cannot be removed with whole bowel irrigation. Although hemodialysis does not enhance elimination of iron, it may be necessary to remove the ferrioxamine complex in patients with renal failure.
Table 120-6. Estimation of Iron-Poisoning Severity
ISONIAZID
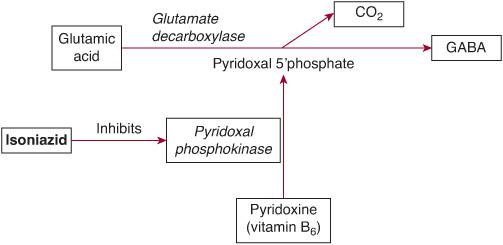
Isoniazid (INH) is a first-line antituberculous medication and is a hydrazide derivative of isonicotinic acid. Acute INH toxicity causes deficiency of gamma-aminobutyric acid (GABA) in the central nervous system, resulting in seizures and coma (Fig. 120-3). Metabolites of INH deplete pyridoxine (vitamin B6) and prevent conversion to pyridoxal 5′-phosphate, which is an essential cofactor for the conversion of glutamate (excitatory neurotransmitter) to GABA (inhibitory neurotransmitter). INH also inhibits the conversion of lactate to pyruvate, exacerbating the lactic acidosis resulting from seizures.
Vomiting, ataxia, respiratory depression, coma, and seizures occur within 2 hours of acute isoniazid overdose. Severe anion gap metabolic acidosis develops after seizures, but does not typically occur in their absence. Rhabdomyolysis often results from prolonged seizures. Elevated liver enzymes may occur within several days.
Isoniazid poisoning should be suspected in an unknown overdose with the characteristic presentation of coma, unexplained seizures (especially if they are refractory to standard therapy), and profound metabolic acidosis. Arterial blood gas analysis, electrolytes, blood urea nitrogen, creatinine, calcium, lactate, and glucose are needed to evaluate for other etiologies of seizures and elevated anion gap metabolic acidosis.
Initial management consists of standard supportive care for seizures, coma, and metabolic acidosis. Benzodiazepines alone may be ineffective in terminating isoniazid-induced seizures. The specific antidote is pyridoxine (vitamin B6), which restores the ability to synthesize gamma-aminobutyric acid (GABA). Pyridoxine and benzodiazepines act synergistically because pyridoxine reverses the GABA deficiency, while benzodiazepines sensitize postsynaptic receptors to GABA. The dose of pyridoxine is 70 mg/kg intravenously in children up to a maximum of 5 g in adults, infused at a rate of 1 g per minute until seizures are controlled. If seizures are terminated before the full dose of pyridoxine is administered, the remainder may be infused over the next 4 hours while the isoniazid is gradually cleared. If seizures recur, the dose of pyridoxine may be repeated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


