Thyroid
INTRODUCTION
Disorders of the thyroid are one of the most common reasons for pediatric endocrinology consultations in the newborn period. Physical symptoms of thyroid disease are often absent, but the effects of thyroid dysfunction in the neonatal period could result in significant deleterious health effects later in life. This chapter reviews disorders of the thyroid most likely to be encountered in the newborn nursery and neonatal intensive care unit (NICU).
THYROID DEVELOPMENT AND FUNCTION
Prenatal Development of the Hypothalamic-Pituitary-Thyroid Axis
The hypothalamic-pituitary-thyroid system develops during the first trimester of gestation. The hypothalamus forms from the neural plate by gestational week 6.1 The anterior pituitary is derived from oral ectodermal tissue that becomes Rathke’s pouch. Studies have demonstrated formation of the pouch as early as 8.5 days and separation from the ectodermal tissue by 12.5 days of gestation.2,3 Several transcription factors are responsible for formation of the anterior pituitary cells, including HESX1, sonic hedgehog (SHH), SIX3, ZIC1, and others. HESX1 mutations have been reported in septo-optic dysplasia, which results in pituitary hypoplasia.4 Mutations in SHH, SIX3, and ZIC1 are associated with holoprosencephaly.5–7 Pit1 is a major transcription factor responsible for formation of thyrotrophs (thyroid-stimulating hormone-producing cells), somatotrophs (growth hormone-producing cells), and lactotrophs (prolactin-producing cells).8 This factor interacts with the promoter regions of genes responsible for production of the β subunit of thyroid-stimulating hormone (TSH) and the thyrotropin-releasing hormone (TRH) receptor and is involved in thyroid hormone receptor formation.9,10
The thyroid gland is derived from components of the pharyngeal floor and the fourth pharyngobronchial pouch.1 Follicular cells migrate from the pharyngeal floor along the thyroglossal duct until they merge with the parafollicular C cells formed from the fourth pharyngobronchial pouch. The thyroid is fully formed and located in its normal position in the anterior neck by 50 days’ gestation.1,11 Morphologic errors that occur during these 50 days can result in ectopic thyroid tissue anywhere along the migratory tract, including sublingually, high in the neck, or even in the mediastinum or the heart.1 Rarely, thyroid tissue may be found in a thyroglossal duct cyst, when the thyroid does not migrate appropriately and the thyroglossal duct simultaneously does not appropriately degenerate.
Multiple homeobox genes and transcription factors are involved in normal development and migration of the thyroid. Thyroid transcription factor 1 (TTF-1 or Nkx2.1) is necessary for survival and proliferation of primitive thyroid cells.1 Thyroid transcription factor 2 (TTF-2 or FOXE-1) is responsible for proper migration of thyroid tissue to its normal position in the neck.12,13 Pax8 is important for follicular cell development.13,14 Other genes, such as various Hox, Hex, FOXE-2, and Eya subtypes, interact with TTF-1, TTF-2, and Pax8 to form the thyroid gland.13 Although critically important in thyroid development, mutations in TTF-1, TTF-2, and Pax8 genes are responsible for only 2% of thyroid dysgenesis in newborns with congenital hypothyroidism (CH).14
Maturation of the Fetal Thyroid
The fetal thyroid gland begins to function at 14 weeks’ gestation, which is when it is able to trap iodine. Thyroid hormone production is minimal until 18–20 weeks’ gestation, when iodine uptake is increased and thyroxine (T4) levels rise throughout the remainder of gestation.15–18 Triiodothyronine (T3) concentrations remain low until about 30 weeks’ gestation and then rise slowly until term. This is due in part to low tissue concentrations of type I iodothyronine monodeiodinase, which converts T4 to T3, and high concentrations of type III monodeiodinase in the placenta and many fetal tissues, which converts T4 to the inactive reverse T3 (rT3). As the fetal liver matures, hepatic type I monodeiodinase increases T4 to T3 conversion, resulting in rising T3 levels.19
Pituitary secretion of TSH begins in the 14th week of gestation. TSH concentrations begin to rise significantly between 16 and 18 weeks’ gestation until a TSH surge occurs immediately after birth.20 The pituitary begins responding to TRH by 25 weeks’ gestation. Pituitary production of TSH seems to initially occur independently of T4 production, as the fetal TSH concentration continues to rise throughout gestation despite normal adult concentrations of T4 by the third trimester.18 However, the ratio of TSH to free T4 progressively decreases into extrauterine life, suggesting that the negative-feedback system matures as TSH-producing cells in the pituitary become more sensitive to circulating T4 concentrations.21
Thyroid Hormone Metabolism and Transport
Eighty percent of T3 found in the body is derived from T4 that undergoes deiodination in peripheral tissues.22 T4 is produced exclusively by the thyroid gland. The peripheral conversion of T4 to T3 is controlled by 2 enzymes, type I and type II monodeiodinase.23,24 Type I monodeiodinase is found predominantly in the liver, kidney, and thyroid itself, whereas type II monodeiodinase is found in the brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissue. Type I monodeiodinase can be inhibited by propylthiouracil (PTU), whereas type II cannot.1,23 Type II monodeiodinase is the major enzyme involved in peripheral conversion of T4 to T3.22–24 It is particularly important in making sure the brain is provided with sufficient T3 for development.25,26
The thyroid hormones can also be irreversibly inactivated by deiodination of T4 and T3 into rT3 and diiodothyronine (T2), respectively, which are biologically inactive.22 This inactivation can be catalyzed by type I monodeiodinase but is mostly controlled by a third enzyme, type III monodeiodinase. This enzyme is found in the brain, placenta, pregnant uterus, and most fetal tissues.27 Type II and type III deiodinase work to regulate the amount of active thyroid hormone to which the fetus is exposed from the mother.27,28 All 3 enzymes are regulated by the thyroid status of the fetus.1
T3 and T4 travel in the blood bound to proteins, primarily thyroid-binding globulin (TBG). Forty-nine to 64% of total T4 and 80% of total T3 are bound to TBG.29 Fetal TBG production by the liver increases in the second half of gestation and is stimulated by placental estrogen.23 TBG and other transport proteins are discussed in greater detail further in the chapter (in the section on disorders of thyroid hormone transport proteins).
At the time of birth in full-term infants, there is a large increase in TRH and TSH that results in the so-called TSH surge. TSH levels peak to as high as 100 mU/L by about 30 minutes after birth and then decrease progressively to normal levels by 3 to 5 days of life.21,30,31 T4 also rises, in response to this surge, and free T4 levels remain elevated for up to 10 weeks after birth.32 T3 levels also rise due to increased thyroidal production in response to the surge and increased activity of hepatic type 1 monodiodinase.28,33
Placental Iodine and Thyroid Hormone Transport
The biologically active thyroid hormones T3 and T4 are formed by coupling of tyrosine rings and organification of iodide. The placenta transports iodide to the fetus via a Na+/I− symporter.34,35 The iodide can then be used to synthesize thyroid hormones. Severe CH occurs in populations of mothers with insufficient iodine intake and may affect the brain development in the fetus.13,36,37 The mechanism for hypothyroidism is 2-fold. In the first trimester, prior to the fetal thyroid gland functioning, the fetus is dependent on maternal T4. When maternal iodine intake is insufficient, she becomes hypothyroid, and the fetus does not have sufficient T4. The second is maternal and fetal thyroid competition for iodide.38 When iodide concentrations are low in the maternal serum, the fetal thyroid is unable to increase its affinity for iodide despite upregulation of the Na+/I− symporter, resulting in underproduction of fetal T4.32,34
Influence of Maternal Thyroid Function on the Fetus
While the fetal thyroid begins to function at 14 weeks’ gestation, the fetal brain T3 receptors are detectable by 10 weeks’ gestation.16,39 The placenta contains monodeiodinases that convert most maternal T4 and T3 to the inactive thyroid hormones rT3 and T2, respectively.13,27 However, T4 and T3 have been found in fetal tissues as early at 9 weeks’ gestation, and by 13 weeks, T3 concentrations in the cortex have been found to be 50%–60% of those found in adults.40 Children born to mothers with undiagnosed hypothyroidism in the first trimester have been found to have cognitive impairment.41–44 Newborns without functional thyroid glands have low levels of circulating T4 at birth of maternal origin.45,46 Presence of circulating maternal T4 in utero is neuroprotective and in the newborn is, at least partially, responsible for the frequently asymptomatic nature of CH early in postnatal life. Figure 47-1 summarizes maternal-placental-fetal interactions with respect to the hypothalamic-pituitary-thyroid axis.47
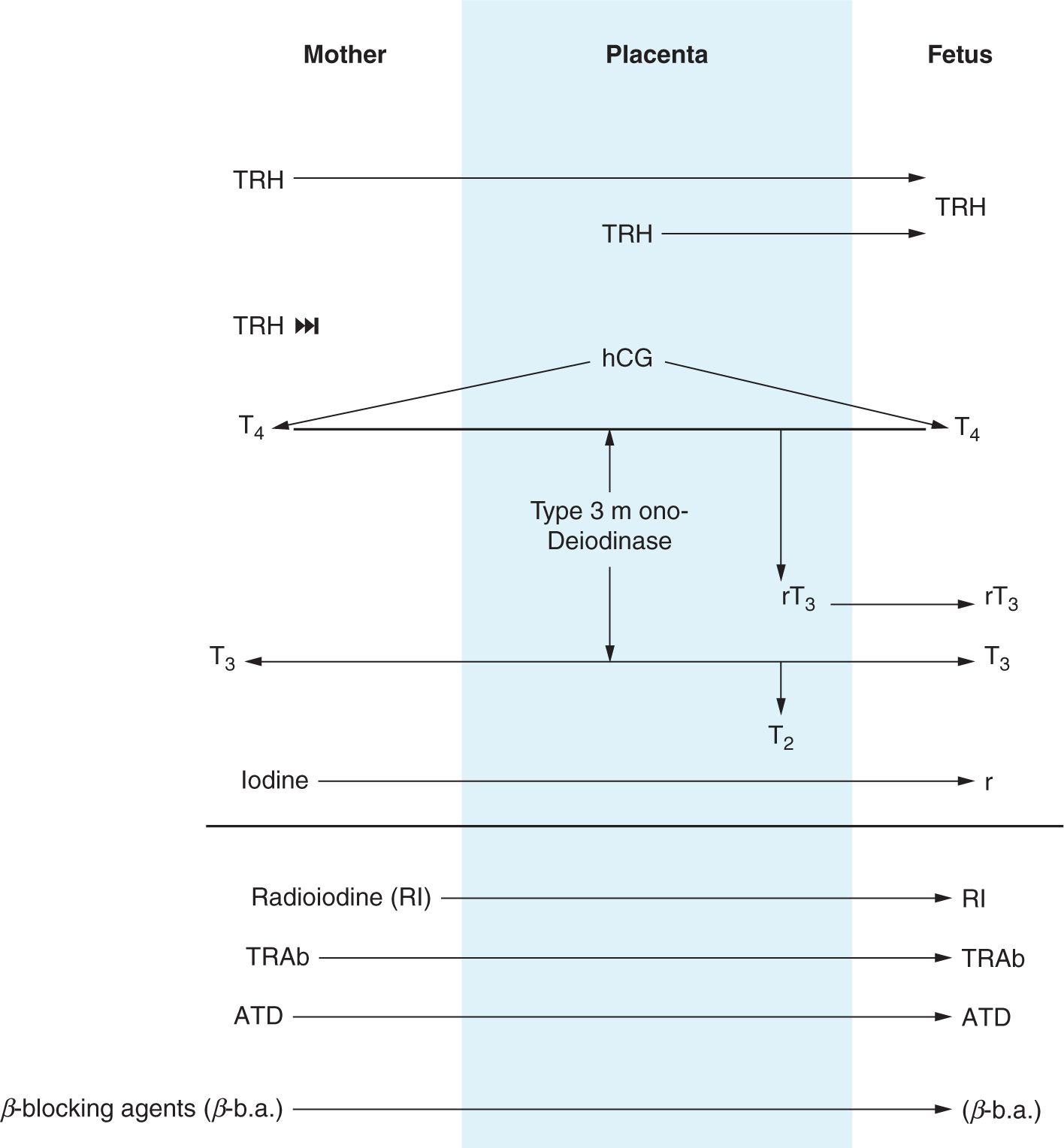
FIGURE 47-1 Maternal-placental-fetal transport of hormones and medications related to the hypothalamic-pituitary-thyroid axis. ATD, antithyroid drugs; β-b.a., β-blocking agents; hCG, human chorionic gonadotropin; I, iodine; RI, radioactive iodine; rT3, reverse triiodothyronine; T2, diiodothyronine; T3, triiodothyronine; T4, thyroxine; TRAb, thyrotropin receptor antibodies; TRH, thyrotropin-releasing hormone; TSH, thyrotropin. (Adapted from Peter and Muzsnai.47)
DISORDERS OF THYROID HORMONE TRANSPORT PROTEINS
The majority of thyroid hormone, present in serum, is bound to proteins, including thyroid TBG, albumin, and T4-binding prealbumin, also known as transthyretin (TTR).48 Of these proteins, TBG carries the majority of T4 and T3.49 In general, patients with defects of thyroid hormone transport proteins are euthyroid, with normal measurement of free T4 and TSH, but have measured abnormalities in the total T4.
Both complete and partial TBG deficiency have been described. TBG deficiency is X linked; consequently, it is more common in male infants. However, due to variable X inactivation, female infants can also express TBG deficiency. Prevalence estimates range from 1 in 5000 to 1 in 12,000 newborns.1
Excess of TBG, which is also X linked, is less common, occurring in 1 in 6000 (Birmingham, UK) to 1 in 40,000 (New York, USA) newborns on screening. The variability in the reported prevalence may be related to transient TBG excess. Those with inherited forms of TBG excess have levels that are 3 to 4.5 times the normal range in affected males and intermediate between normal and affected males in female carriers.50
Familial dysalbuminemic hyperthyroxinemia (FDH), which is inherited in an autosomal dominant fashion, is the most common cause of euthyroid hyperthyroxinemia, occurring in up to 1.8% of Hispanics but thought to be less common in other ethnic groups.48,51 Individuals with FDH are clinically euthyroid with elevated total T4 levels.
Rarely, TTR variants have been described. Autosomal dominant mutations in the TTR gene have been described that increase the affinity for T4 and lead to detection of low levels of total T4 in the serum in otherwise-euthyroid individuals.52 Likewise, mutations of the TTR gene that decrease the affinity for T4 and lead to detection of elevated levels of total T4 in the serum in otherwise-euthyroid individuals have been described. Individuals who are homozygous and heterozygous for TTR mutations have been reported.53
MEDICATIONS AFFECTING THYROID FUNCTION
Although uncommon in the United States, iodine deficiency remains the most common cause of hypothyroidism worldwide and can have an impact on both a pregnant mother and her fetus. Somewhat paradoxically, iodine excess also acutely leads to hypothyroidism via the Wolff-Chaikoff effect, presumably as a feedback mechanism to prevent hyperthyroidism. High levels of dietary iodine, for example in Asian cultures with heavy consumption of seaweed, have been associated with both hypothyroidism and hyperthyroidism.54 Rare cases of CH related to excessive iodine intake in nutritional supplements by a pregnant mother have also been reported.55 Transient hypothyroidism from topical iodine exposure has been reported and should be in the differential diagnosis but is not a common cause of transient neonatal CH in North America.56 The premature infant is at particularly high risk because of the increased frequency of procedures and the immature skin that allows increased absorption. Although there is the potential for iodinated contrast during computed tomographic (CT) studies also to negatively affect the developing thyroid gland of the fetus, a retrospective study of 21 pregnancies in which contrast CT studies were performed between 8 and 37 weeks’ gestation (mean 23) revealed no evidence of hypothyroidism in the offspring.57
Dopamine and glucocorticoids interfere with TSH release from the pituitary and therefore could contribute to central hypothyroidism or mask TSH rise in primary CH. Antithyroid medications used in Graves disease are discussed further in the chapter (in the neonatal hyperthyroidism section).
Several other drugs are well known to have an impact on the thyroid axis; however, many are not routinely utilized in pregnancy or the neonatal period. For example, lithium is associated with thyroid dysfunction; however, it is a class D drug in pregnancy because of its link with congenital heart disease, and there are no indications for its use in neonates. The antiarrhythmic amiodarone alters thyroid hormone secretion but is uncommonly used in pregnant woman and neonates. Transient hypothyroidism has been reported in infants born to mothers treated with amiodarone.58 The uses of other medications, such as interferon α, in pregnancy are at the case report level.59
NEONATAL HYPOTHYROIDISM
Congenital hypothyroidism is a common preventable cause of mental retardation. Newborn screening programs for CH have been a huge public health success, such that severe mental retardation as a consequence of CH has been essentially eliminated.
Primary Congenital Hypothyroidism
Epidemiology
Prior to the onset of newborn screening, the incidence of primary CH was estimated to be 1 in 7000 births.60 With the advent of newborn screening, the incidence was initially lowered to the 1:3–4000 range.61 More recent data suggest the incidence may be even higher. In the United States, the incidence of primary CH increased from 1:4098 in 1987 to 1:2370 in 2002. In New York State, the reported rates have been even higher,62 with 1:2278 in 1978 to 1:1414 in 2005. The near doubling of primary CH over the past 2–3 decades is not limited to the United States.63 Several proposed reasons likely together contribute to the increasing incidence of primary CH, including changing trends in ethnic populations, earlier screening, higher proportion of low birth weight infants, and possibly additional prenatal environmental exposures.64,65 Perhaps most significantly, lower TSH cutoffs have increased identification of mild hypothyroidism.66,67
Higher incidences of primary CH have been reported in Hispanic, Asian, and Native American ethnic groups, with lower incidences among whites and non-Hispanic blacks.63,64,68 There is an unexplained higher incidence of females with primary CH. An odds ratio of 1.56 was reported in the United States from 1991 to 2000. The sex-specific odds ratio is increased in certain ethnic groups and has been reported as high as 2.6 in Hispanic newborns in California.64
Pathophysiology and Genetics
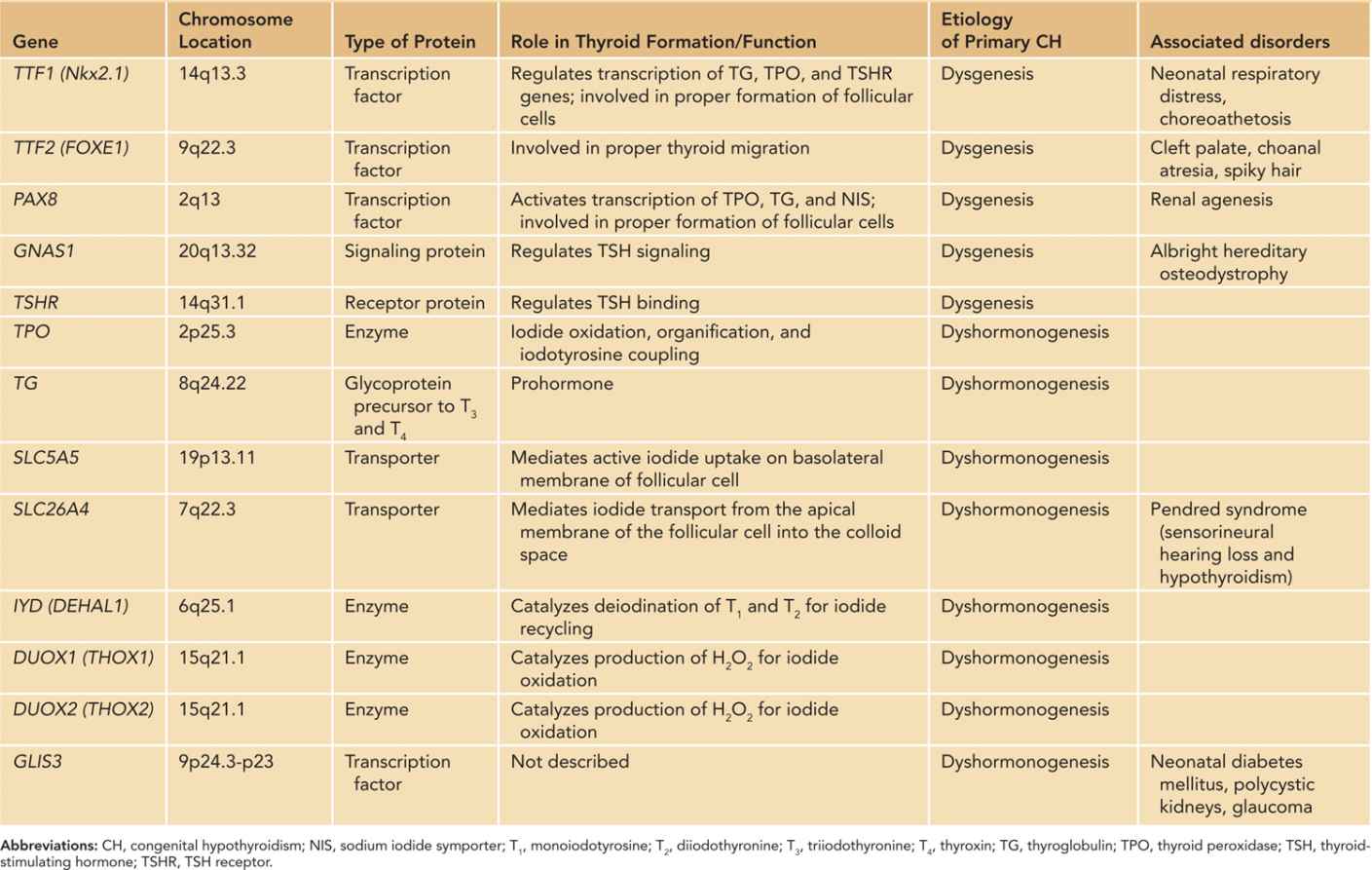
Congenital hypothyroidism can result from several distinct pathophysiological mechanisms. Primary hypothyroidism describes the condition in which there is inadequate thyroid hormone synthesis or release in response to TSH, leading to the distinctive laboratory feature of elevated TSH. The 2 major causes of primary hypothyroidism are thyroid dysgenesis, in which the thyroid gland does not properly form, and dyshormonogenesis, when the gland forms normally and in the correct location but does not function appropriately. Together, they make up the majority of primary CH cases.69 Table 47-1 summarizes the known causative genes associated with primary CH.47,70–72
Table 47-1 Genes Associated With Congenital Primary Hypothyroidism
Thyroid Dysgenesis
Developmental disorders of the thyroid ranging from complete absence of thyroid gland development to hypoplasia to ectopic placement historically make up the majority of cases of primary CH. Thyroid dysgenesis is typically considered sporadic, such that the risk of recurrence in a future pregnancy is only modestly different from the general population. Familial cases have been shown to occur in 2%–3% of cases, and causative mutations in genes encoding transcription factors critical to thyroid development and differentiation, including PAX-8, TTF-1, TTF-2, and GL-153, have been identified.71,73–75 While thyroid dysplasia has been estimated to constitute approximately 75% of CH cases, this may no longer hold true with the recent change in incidence driven by increasing identification of mild cases. An Italian study identified 435 cases of CH with a cutoff of 10 mU/L over a 7-year period, calculated as an incidence of 1:1446. They then grouped the cases by etiologies, athyreosis/profound hypoplasia, ectopy/hemiagenesis, or gland in situ and determined that 68% were classified as gland in situ, that is, not thyroid dysgenesis, conflicting with the long-held postulate about frequency of different etiologies of CH.76
Dyshormonogenesis
Disorders in which the thyroid gland fails to concentrate iodide, form thyroid hormone, and secrete it into the circulation collectively are referred to as thyroid dyshormonogenesis. Key steps in thyroid hormone synthesis and secretion have been reviewed previously; defects in any of these steps can lead to CH. Included among these are defects of the TSH receptor, sodium/iodide symporter, peroxidase system, thyroglobulin, and deiodinases.77
Transient and Mild Hypothyroidism
While thyroid dysgenesis and dyshormonogenesis are considered to have established pathophysiological mechanisms, the diagnoses of transient and mild hypothyroidism are somewhat controversial. Transient elevations in TSH can have identifiable etiologies, such as maternal antithyroid drugs, maternal thyrotropin receptor-blocking antibodies (TRAbs), or exposure to excessive iodine.78,79 Genetic etiologies have rarely been described, but it is known that patients heterozygous for DUOX2 and THOX2 mutations can have transient or mild CH.80,81 Most often, the cause of transient CH is not definitively determined. In addition, as one cannot always predict with certainty whether an infant has permanent or transient hypothyroidism, clinicians will often begin treatment in the neonatal period, thereby precluding the opportunity to observe the natural history. Meanwhile, the cutoff TSH value for upper range of normal is arbitrary, and no single feature reliably consistently distinguishes mild hypothyroidism from normal physiology.
Controversy exists regarding whether to treat infants with “borderline” or “subclinical” hypothyroidism: those with normal T4 but TSH levels persistently above the upper limit of normal but less than 10 μIU/mL. As TSH is considered the most sensitive indicator for hypothyroidism, it is possible that the mild elevation is an indicator of insufficient T4, even if the T4 is within the normal reference range for the assay. There are no controlled studies in otherwise-healthy infants to evaluate outcomes in those treated with levothyroxine (L-T4) for mild TSH elevations vs those left untreated. A randomized, double-blind, placebo-controlled study of children with Down syndrome and borderline TSH values in the newborn period showed that those who received L-T4 in the first 2 years of life had better motor development and a trend toward, but not significantly, better cognitive development. The L-T4-treated group also had a better length at 2 years of age when compared to the placebo-treated group, suggesting that treatment may benefit growth.82 TRH tests have been used to identify infants with mild elevations of TSH who are most likely to benefit from treatment.83
Newborn Screening
Newborn screening for CH to prevent the outcome of mental retardation fits Wilson and Jungener’s 1968 classical principles for screening for a medical condition in every respect.84
Infants born with CH may be indistinguishable from those without CH on physical examination; however, a small sample of blood obtained by heel stick is sufficient to identify the condition. The most significantly affected newborn will achieve normal or near-normal cognitive outcomes when treatment is initiated early. Newborn screening for CH first became available in the 1970s with pilot programs originating in Quebec, Toronto, and Pittsburgh, Pennsylvania.85 Since then, newborn screening for CH has been established as a mainstay of newborn care in the United States and the industrialized world.
Primary T4 Screening
Using filter paper blood spots, an initially low or low-normal T4 measurement is followed up with a TSH measurement. A major advantage of the primary T4 screening test is that it has the potential to detect infants with both primary and central hypothyroidism. In addition, it will detect infants with TBG deficiency, and if high values are reported, T4 screening can detect hyperthyroxinemia. A potential challenge with interpretation is with infants who have an initially normal T4 concentration, especially if samples are collected close to birth, when the T4 surge may give a false-negative result. For this reason, special consideration should be given to children with Down syndrome in regions with primary T4 screens and a TSH sample should be obtained in the newborn period given the high incidence of primary CH in this population of children.86 T4 screening is also associated with a higher false-positive rate, generating additional recall visits.61
Primary TSH Screening
As the primary condition to identify by newborn screen is primary hypothyroidism, for which the most sensitive laboratory finding is hyperthyrotropinemia, it follows that screening by TSH has significant appeal. With improvement in the TSH assay on dried spot filter paper samples coupled with the recognition that a primary TSH screen has a lower false-positive rate than primary T4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


