Thymic Aplasia and T-Cell Disorder
INTRODUCTION
As detailed in chapter 49, host defense mechanisms against pathogens are defective in the newborn as a result of immature immunity. Preterm infants are particularly vulnerable to infections associated with prematurity-related complications and medical interventions. The recognition of inborn errors of immunity and differentiation from functional immaturity of the immune system pose a diagnostic challenge. An organized clinical approach is essential to facilitate early diagnosis, which has a significant impact on prognosis and survival.
To date, more than 150 monogenic primary immunodeficiency disorders (PIDs) have been described.1 They can be classified into (1) combined T- and B-cell immunodeficiencies, (2) predominantly antibody deficiencies, (3) immune dysregulatory syndromes, (4) phagocytic disorders, (5) defects in innate immunity, (6) diseases of immune dysregulation, (7) complement deficiencies, and (8) other well-defined disorders, such as Wiskott-Aldrich syndrome (WAS) and DNA repair defects. Among them, combined T- and B-cell deficiencies constitute the most clinically important group of disorders in neonates and infants. The lack of cell-mediated immunity results in susceptibility to opportunistic infections, which is the principle manifestation leading to diagnosis in most infants with severe combined immunodeficiency (SCID). In addition, defective T-cell homeostasis may lead to immune dysregulation and lymphoproliferation, which are less commonly encountered in the neonatal period.
EPIDEMIOLOGY
Primary immunodeficiency disorders are rare disorders. SCID is estimated to occur in 1:50,000 to 1:100,000 live births, but the exact incidence is unknown as some affected infants might have died before a diagnosis could be made.2 In addition, atypical SCID caused by hypomorphic mutations may be underdiagnosed. The implementation of newborn screening (NBS) for SCID is expected to provide epidemiological data on the true incidence rate.
The spectrum of immunological phenotypes and genetic etiology vary with ethnic background and consanguinity rate of the population. In the United States, interleukin (IL) 2 receptor γ chain (γc) deficiency of T−B+ immunophenotype inherited in an X-linked manner accounts for 45%–50% of SCID.2 However, T−B−SCID of autosomal recessive (AR) inheritance predominates in the Athabascan-speaking Navajo and Apache native Americans because of the high frequency of DCLRE1C (Artemis) founder mutation (2.1%), giving rise to an incidence as high as 1:2000 live births in these ethnic groups.3 AR-SCIDs are also more common in populations with a high prevalence of consanguineous marriage, such as North Africa and the Middle East.4–6
Other types of T-cell disorders presenting in the neonatal and infancy period, such as WAS and familial hemophagocytic lymphohistiocytosis, are much rarer, and the incidence varies from 1:100,000 to 1:1,000,000.7,8
PATHOPHYSIOLOGY
The immune response is a complex host defense network that functions to protect the body against pathogens, recognize self- and non-self-antigens, sense DNA damage, and eliminate malignant clones. To achieve these tasks, the system requires a functional repertoire consisting of optimal numbers of immune cells that can generate specific immune response to a diversity of pathogens, effectively remove the pathogen, and elicit recall memory response. The immune system must also recognize and reject alloantigens while sustaining immune tolerance to self-antigens and harmless environmental antigens. Finally, effective immune surveillance toward DNA damage is required to prevent malignancy. The immune response is tightly regulated to maintain homeostasis and prevent tissue damage from overreactivity.
Disorders of Lymphocyte Development and Differentiation
Disorders of T-cell development and differentiation constitute the most important category of T-cell disorder in neonates and infants. Molecular defects leading to numerical and functional T-cell deficiencies can be classified into 6 main categories (Table 50-1).9
Table 50-1 Genetic Etiology and Immunophenotype of Severe T-Cell Defects Manifesting in Neonatal or Early Infancy Period
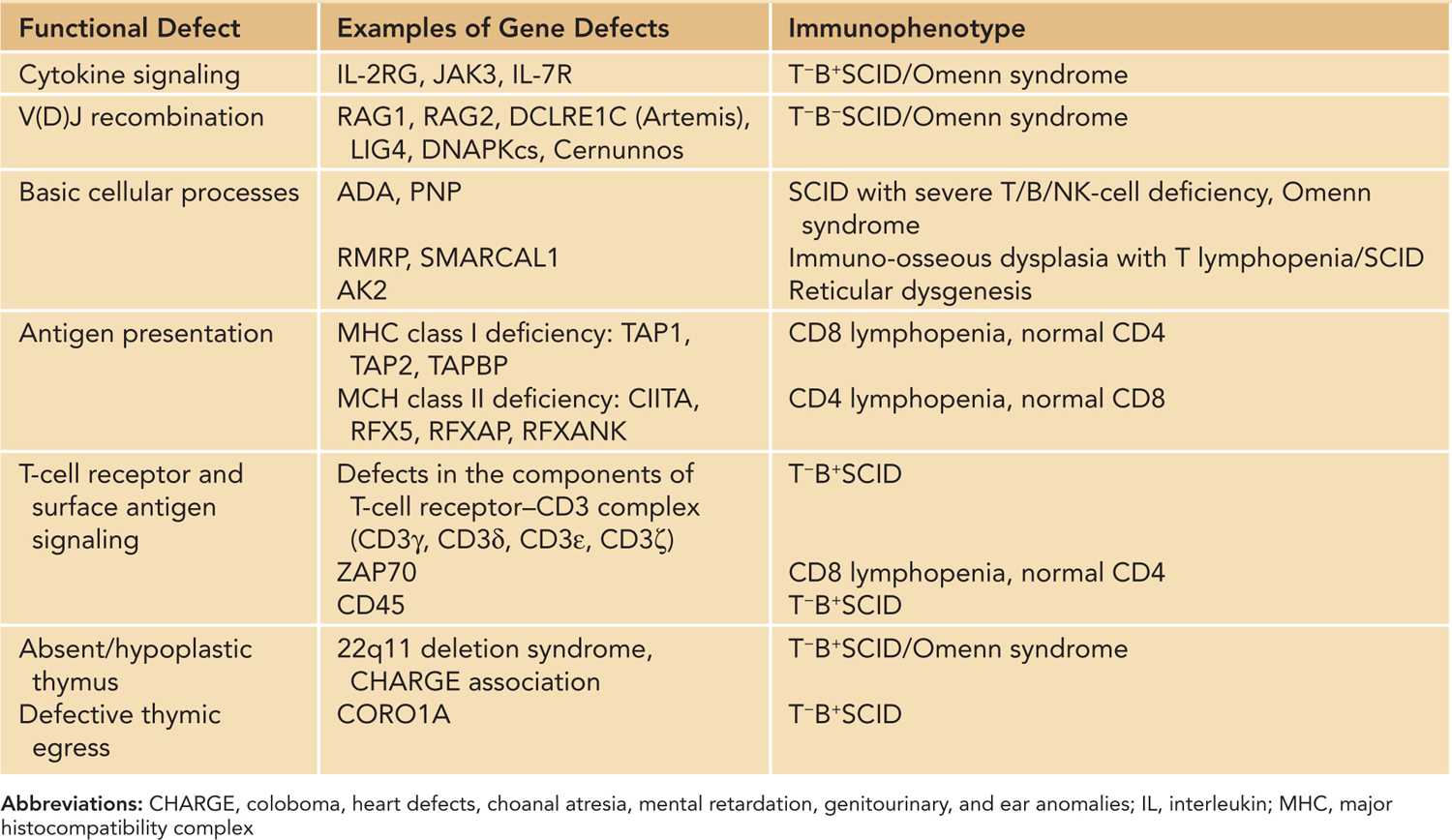
Cytokine Signaling
The development, proliferation, survival, and differentiation of T cells take place in the thymus and are supported by cytokines, including IL-2, IL-4, IL-7, IL-9, and IL-15.10 Receptors of these cytokines share γ c as a common component. Cytokines bind to their receptors and activate the JAK-STAT signaling pathway, leading to transcription of specific genes. IL-2 promotes T-cell growth, immunoglobulin (Ig) production, and natural killer (NK) cell cytotoxicity. Therefore, SCID caused by defects in γ c and JAK3 are characterized by severe depletion of T cells and NK cells. The B cells are present but are functionally abnormal due to the lack of T-cell help and defective IL-4- and IL-21-mediated signaling. In IL-7 receptor α -chain (IL7Rα) deficiency, T cells are absent but B cells and NK cells are present.11
V(D)J Recombination
Normal maturation of T cells and B cells depends on successful somatic recombination of variable (V), diversity (D), and joining (J) gene segments that encode the variable regions of the T-cell receptors (TCRs) and immunoglobulin receptors, respectively. V(D)J recombination is initiated by the introduction of site-specific DNA double-strand breaks (DSBs) to regions flanking the V, D, and J gene segments, and the broken ends are rejoined by the nonhomologous end-joining (NHEJ) pathway. V(D)J recombination defects lead to a block of T-cell and B-cell maturation, while NK lineage is spared, giving rise to a T−B−NK+ immunophenotype. The ubiquitously expressed NHEJ pathway is also an important DNA repair mechanism for maintaining genomic integrity; hence, tissue cells with dysfunctional NHEJ DSB repair machinery are prone to mutagenesis induced by ionizing radiation.12
Defects in Basic Cellular Processes
A number of PIDs are caused by disorders in various housekeeping processes, such as purine salvage pathways and ribosome biogenesis. Adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) are enzymes of the purine salvage pathway expressed in all tissues of the body, with the highest expression in lymphoid cells. Purine metabolites are toxic to the lymphocyte progenitors, and they interfere with V(D)J recombination. Apoptosis of the immature thymocytes results in profound lymphopenia.13 Both ADA and PNP deficiencies are associated with neurological deficits, such as developmental delay, learning disability, and sensorineural hearing loss.14 ADA deficiency has additional organ involvement, including pulmonary interstitial inflammation, skeletal abnormalities, and hepatic and renal dysfunction.
Dysfunction of ribosomal biogenesis accounts for several congenital bone marrow failure syndromes, among which cartilage-hair hypoplasia (CHH) exhibits severe T-cell defects. CHH is caused by mutation of the ribonuclease mitochondrial RNA-processing endonuclease (RMRP), which is involved in ribosomal RNA cleavage. Affected neonates displays short-limb dwarfism related to underlying metaphyseal chondrodysplasia. The immunological defect is thought to be related to disturbed cell cycle control.15
Reticular dysgenesis is caused by a defect of adenylate kinase 2, a mitochondrial enzyme involved in cellular energy generation by oxidative phosphorylation. It manifests early in the neonatal period with SCID and profound neutropenia as a result of developmental arrest in myeloid and lymphoid lineages, especially T and NK cells. Affected infants also have severe sensorineural deafness.16,17
Defect of T-Cell Receptor Signaling
The response of T lymphocytes to antigens is mediated through signaling via the antigen receptor complex, which consists of the TCRαβ or TCRγδ and the associated invariant accessory proteins collectively called CD3 (CD3δε, CD3γε, CD3ζζ). Signaling through the multimeric TCR/CD3 complex determines the development, survival, and effector functions of T lymphocytes.18 CD3δ, CD3ε, CD3ζ deficiencies are known to cause SCID with T−B+NK+ phenotype. CD3γ deficiency leads to partial T lymphopenia, and clinical severity is more variable.19,20 Deficiency of other molecules involved in TCR signaling, such as CD4521 and ζ -associated protein, 70kd (ZAP70), were also reported in young infants with SCID manifestations. Characteristically, patients with ZAP70 deficiency exhibit very low or total absence of CD8+ T-cells.22
Defects of Antigen Presentation
The CD8+ T lymphocytes recognize viral particles associated with major histocompatibility complex (MHC) class I molecules and CD4+ T lymphocytes recognize antigens presented by MHC class II molecules. Deficiencies of MHC class I or class II molecules are collectively known as bare lymphocyte syndrome.23 Patients with MHC class II deficiency present in infancy with protracted opportunistic infections as they fail to mount CD4+ T-cell-mediated response to specific pathogens and display severe CD4+ lymphopenia and impaired humoral immunity.24 Unlike MHC class II deficiency, the age of onset of MHC class I deficiency varies from childhood to adulthood, and clinical features include recurrent sinopulmonary bacterial infections, bronchiectasis, and granulomatous skin inflammations.25
Disorder of Thymic Development and Functions
The thymus provides an architecturally and functionally organized microenvironment for T-cell development. Immature thymocytes undergo successive stages of TCR gene rearrangement, positive selection, lineage commitment to single-positive (CD4 or CD8) T cells, negative selection, and finally exit the thymus to the circulation as peripheral T-cell repertoire.26 T-cell deficiency occurs in developmental defects of the thymus, such as 22q11.2 deletion syndrome (DiGeorge syndrome, DGS) and CHARGE (coloboma, heart defects, choanal atresia, mental retardation, genitourinary and ear anomalies) association. Abnormal migration of neural crest cells into pouch ectoderm forms the basis of thymic hypoplasia, hypoparathyroidism, and conotruncal cardiac defects in 22q11.2 deletion. Thymic aplasia (“complete” DGS) occurs in less than 0.5% patients with 22q11.2 deletion and manifests as SCID. The majority have impaired thymic development, leading to variable defects in T-cell numbers, T-regulatory cell function, and central tolerance with increased predisposition to infections and autoimmunity.27,28
Haploinsufficiency of the CHD7 gene was identified as a cause of CHARGE syndrome. CHD7 is expressed in the mesenchyme of the pharyngeal arches, and a small number of patients were found to have thymic aplasia or hypoplasia.29 The prevalence of immunodeficiency in patients with CHARGE syndrome is unknown, but a few cases of T−B+NK+ SCID were reported.30
Immunological Consequence of Defects in T-Cell Development
Profound numerical and functional deficits of T and B lymphocytes severely compromise the host’s defense against infections. If residual protein expression or activity is permitted by hypomorphic mutation, impairment of the adaptive immune response is variable, and some function may be retained. Hypomorphic mutations of SCID-causing genes, such as RAG1, RAG2, Artemis, LIG4, IL2RG, IL7R, ADA, RMRP, CHD7, and 22q11.2 deletion, are known to cause Omenn syndrome, which is characterized by generalized erythroderma, lymphadenopathy, eosinophilia, and increased IgE in addition to severe immunodeficiency.9,31–33 The thymus is dysplastic, but the residual, yet aberrant, thymic function allows maturation of a limited number of T cells, which manage to exit the thymus to the periphery, where they expand on activation by antigens. Therefore, T-cell count may be slightly reduced or even increased, but they exhibit an oligoclonal TCR repertoire and fail to mount effective cell-mediated immune response against pathogens.34 Impaired central tolerance as well as abnormal development and function of T-regulatory cells account for the emergence of autoreactive T cells, which cause uncontrolled tissue inflammation and autoimmunity.35,36
Immune Dysregulatory Disorders
Naturally arising regulatory T cells (CD4+CD25+FOXP3+ Tregs) is a specialized T-cell subset defined by high intracellular levels of Foxp3 protein expression. FOXP3 is a transcription factor required for the development and function of Treg cells, which are critical mediators of peripheral tolerance. Treg cells downregulate activated effector T cells by suppressing their production of proinflammatory cytokines and by secreting inhibitory cytokines such as IL-10 and transforming growth factor β (TGF-β).37 The absence of FOXP3-expressing Tregs leads to immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, which is characterized by multiple organ-specific autoimmunity, such as type I diabetes mellitus, thyroiditis, severe enteropathy and villous atrophy, immune cytopenia, dermatitis, and recurrent infections.38,39
When regulatory mechanisms of natural termination of immune response are disrupted, uncontrolled activation and proliferation of immune cells will cause systemic hyperinflammation. Hemophagocytic lymphohistiocytosis (HLH) is a group of disorders caused by ineffective cytotoxic T-cell and NK-cell response secondary to defects in granule-mediated cytotoxicity. Cytotoxic T cells and NK cells kill infected cells by forming an immunological synapse, through which cytolytic granules containing perforin, granzyme, and other serine-specific proteases are released into the target cells.40 Genetic defects of vesicle transport, priming, docking, fusion, and pore formation at the cytotoxic cell–target cell interface result in familial HLH.41 In the majority of cases, though, HLH is secondary to known triggers, such as infections (especially Epstein-Barr virus), malignancy, rheumatic diseases, and metabolic conditions.42 While infected target cells fail to be cleared, the activated inflammatory cells continue to proliferate and infiltrate various organs, causing massive tissue damage. Cytopenia occurs as activated macrophages spontaneously phagocytose blood cells and platelets. The majority of familial HLH present before 1 year of age, and approximately 10% are symptomatic in the neonatal period.
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of programmed cell death. In normal circumstances, proliferation and activation of lymphocytes after antigenic stimulation is kept in check by the process of activation-induced cell death, which is mediated by the interaction between Fas and Fas ligand expressed on activated lymphocytes. This triggers the intracellular caspase cascade and subsequent proteolysis, DNA degradation, and apoptosis.43,44 Molecular defects along the apoptotic pathway lead to ALPS, which presents as lymphadenopathy, splenomegaly, hepatomegaly, lymphocytosis, and autoimmunity. ALPS of neonatal or prenatal onset is often severe.45,46 In general, most patients with ALPS do not have intrinsic susceptibility to infection. There is an increased risk of malignancy.
CLINICAL INDICATORS OF PRIMARY IMMUNODEFICIENCIES AND DIFFERNTIAL DIAGNOSES
The suspicion of potential primary immunodeficiencies relies on the recognition of clinical pointers, such as protracted sepsis with unusual severity and suboptimal response to appropriate antimicrobials, opportunistic infections, inflammatory manifestations such as skin rash and enterocolitis, and autoimmunity such as cytopenia and endocrinopathies. Initiation of immunological investigations should be considered after thorough clinical review for maternal, peripartum, and neonatal risk factors for infections and the source of infection and rule out secondary causes of immunodeficiencies (Table 50-2). Maternal human immunodeficiency virus (HIV) status should be ascertained, and HIV testing of the infant should be considered as appropriate. Awareness of the early signs and laboratory features that indicate a potential underlying immunodeficiency is crucial to facilitate early diagnosis, minimize infection or immune-mediated damage to vital organs, and optimize transplant outcome.
Table 50-2 Causes of Recurrent Infections and Secondary Immunodeficiencies in Neonates

Opportunistic Infections
Infections commonly occurring in patients with T-cell defects and other categories of PIDs are listed in Table 50-3. An infant with SCID classically presents with cough, tachypnea, wheezing, and oxygen desaturation, often with a history of chronic diarrhea, persistent oral candidiasis, poor feeding, and suboptimal weight gain.47 Chest x-ray often shows diffuse interstitial shadowing or ground glass appearance typical of Pneumocystis jiroveci pneumonia (PCP), and absence of thymic shadow may be evident. Identification of P. jiroveci requires a sample obtained by bronchoalveolar lavage, which is also useful to identify other coexisting pathogens, such as cytomegalovirus (CMV), adenovirus, and bacille Calmette-Guérin (BCG) if vaccinated.48 PCP is regarded as an indicator disease for PIDs and HIV infection, though isolated nursery outbreaks of PCP were reported in resource-poor settings.49
Table 50-3 Common Pathogens in Primary Immunodeficiencies
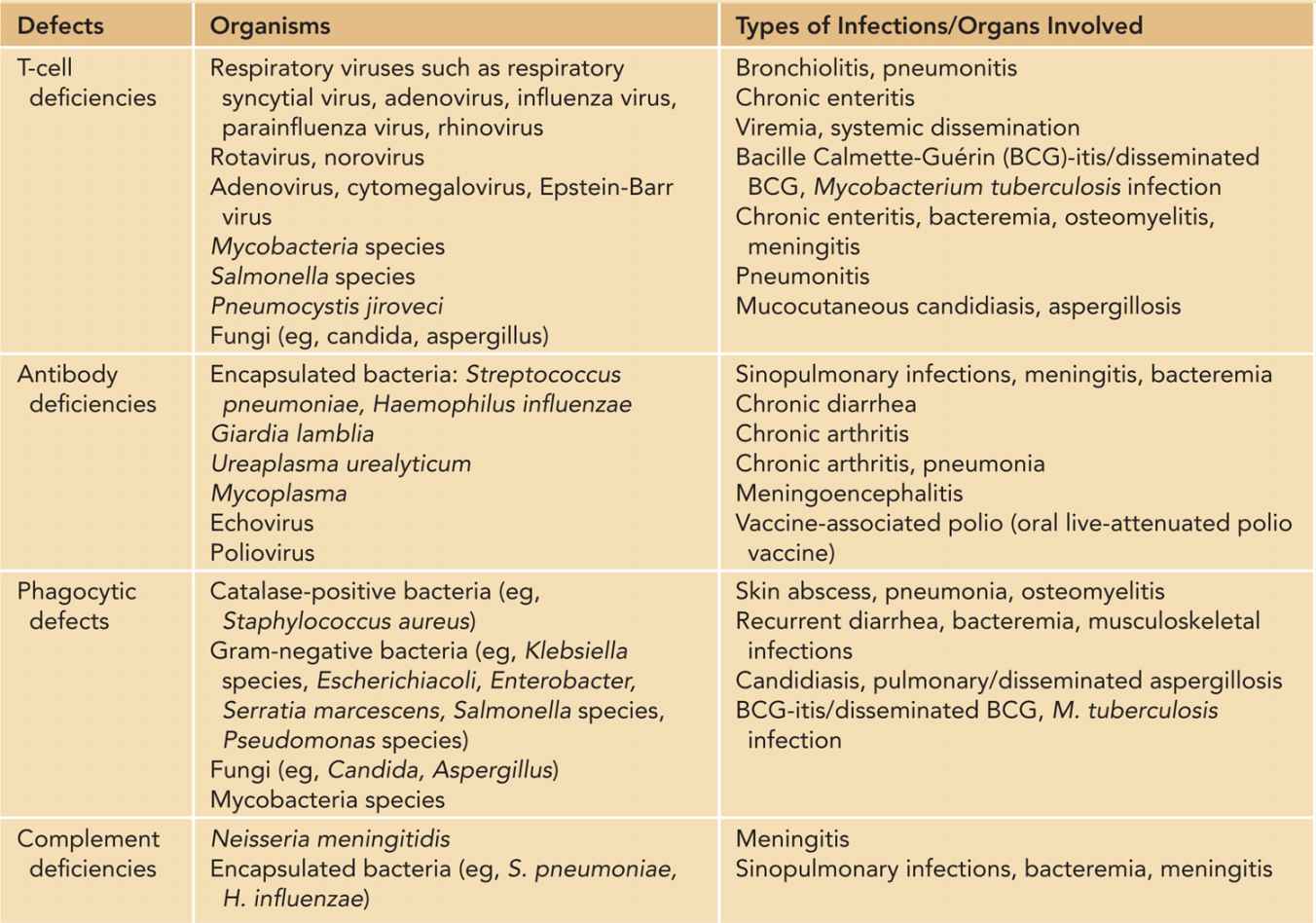
Systemic viral dissemination, such as of CMV and adenovirus, can lead to significant morbidities and mortalities. In addition to bronchopneumonia, infants with disseminated adenovirus infection may develop hepatitis, meningoencephalitis, pancytopenia, and disseminated intravascular coagulation.50 Similar features are also present in disseminated CMV, but chorioretinitis, sensorineural hearing loss, and long-term neurological deficits are special concerns.51 Antiviral agents can provide some degree of control over viral replication, but clearance requires immunoreconstitutive procedures such as hematopoietic stem cell transplantation (HSCT).
Disseminated candidiasis is a frequent infection in infants born under the gestational age of 32 weeks and those with very low birth weight.52 Invasive aspergillosis is uncommon among neonates, but together with candidiasis their incidences have increased since the mid-1990s, coinciding with the increased survival of premature infants.49,52 The occurrence of these fungal infections in infants without obvious risk factors, especially when there are other concurrent opportunistic infections, should raise suspicions for PIDs.
Infants with PIDs undiagnosed at birth may suffer from live vaccine-related illness as they are vaccinated according to schedule. In countries where live-attenuated BCG vaccine is routinely given at birth, regional or disseminated BCG commonly occurs in infants with major primary immunodeficiencies such as SCID, chronic granulomatous disease (CGD), and inborn defects of IL-12-interferon-γ axis.53,54 They present with persistent discharge and ulceration at the BCG inoculation site; ipsilateral axillary lymphadenopathy; and distant and systemic dissemination, including pneumonia, hepatosplenomegaly, multifocal lymphadenopathy, skin nodules, osteomyelitis, and bone marrow involvement. Chronic rotavirus gastroenteritis and disseminated chickenpox has also been reported in infants with undiagnosed SCID who received rotavirus and varicella vaccines.55,56
Skin Eruptions
Infants with various PIDs can present with generalized erythroderma or diffuse exfoliative skin rash. As described in a previous section, erythroderma and secondary alopecia and loss of eyebrows is characteristic of Omenn syndrome, in which activated, oligoclonal T cells infiltrate the skin and elicit an inflammatory response. The skin is thickened and leathery, and skin biopsy typically shows acanthosis and parakeratosis, with an inflammatory infiltrate prominent at the dermis consisting of CD3+ T cells, eosinophils, and some macrophages. Hypoproteinemia and generalized edema often occur because of protein loss through the skin, as well as chronic diarrhea, which often coexists.34
An infant with SCID lacks the ability to reject allogeneic cells. Maternal T lymphocytes are not uncommonly detectable in the infant’s peripheral blood and may cause a graft-vs-host reaction manifesting as diffuse skin erythema, neutropenia, eosinophilia, and increased liver enzymes. The skin eruption resembles atopic dermatitis but preferentially affects the palms and soles.31 Maternofetal engraftment is usually mild or asymptomatic in most cases; in contrast, transfusion of nonirradiated blood products can cause fatal graft-vs-host disease (GVHD) with diffuse necrotizing erythroderma, severe mucosal inflammation of the gut, and biliary tract destruction.57
Other causes of neonatal erythroderma include staphylococcal scalded skin syndrome, congenital cutaneous candidiasis, hereditary ichthyoses such as congenital ichthyosiform erythroderma and harlequin ichthyosis, Netherton syndrome, psoriasis, and drug eruptions. Skin biopsy is helpful to reach the definitive diagnosis.58
Eczematous skin rash also occurs in other forms of PIDs, such as WAS, Job syndrome (autosomal dominant hyper-IgE syndrome), and IPEX.59
Enteropathy
Enteropathy associated with PIDs can be infective, inflammatory, or both. Immunodeficiencies with gastrointestinal symptoms presenting in the neonatal and early infancy period include SCID, CGD, IPEX, ALPS, and defect of NF-κB regulation (NF-κB essential modulator [NEMO] deficiency).60,61 Chronic viral or bacterial enteritis also leads to mucosal damage and villus atrophy. Infants with IPEX may develop autoimmune enteropathy within the first few days of life, and diagnostic clues are provided by evidence of multiorgan autoimmunity, such as type 1 diabetes mellitus and hypothyroidism.62
Congenital Malformations and Dysmorphisms
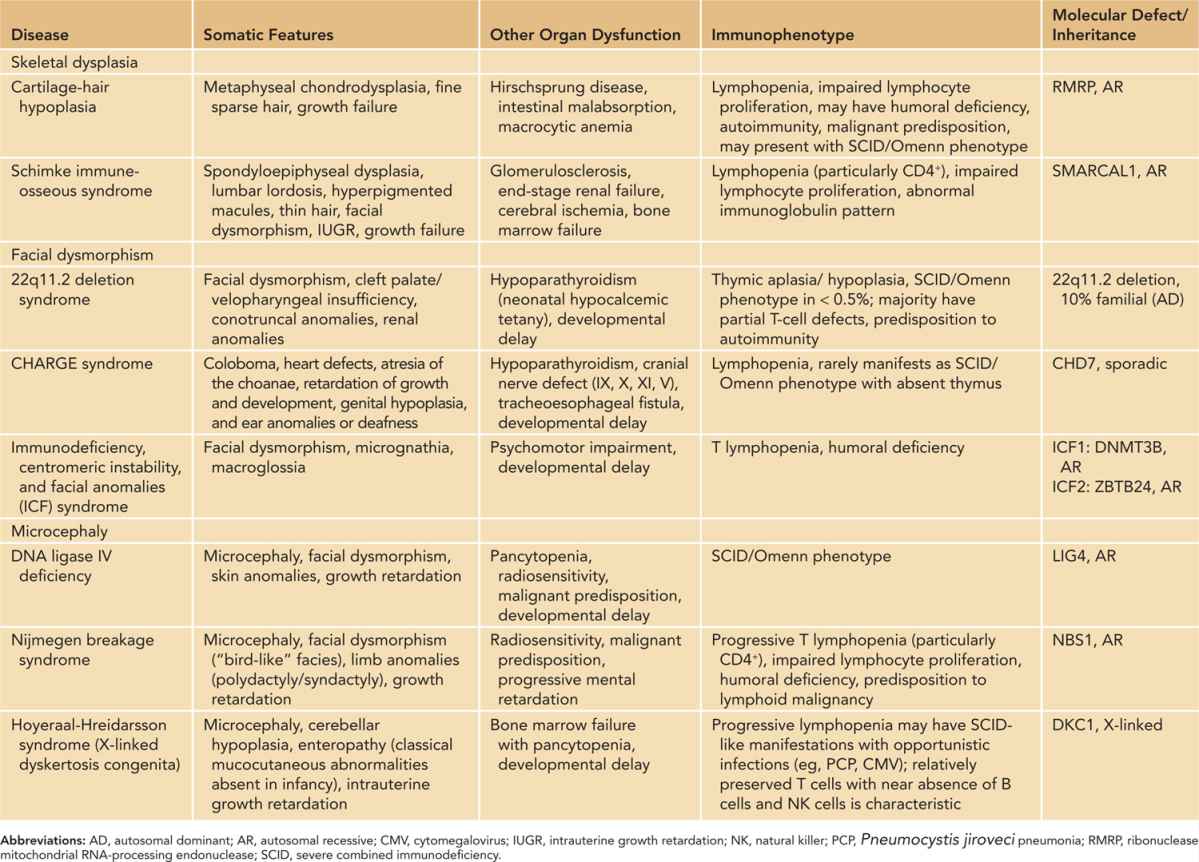
Some forms of PIDs are associated with a characteristic group of structural anomalies or organ dysfunction. In such syndromic PIDs, somatic features are often the presenting manifestations that lead to further investigations. Recognizable syndromes include 22q11.2 deletion syndrome, CHARGE syndrome, immuno-osseous dysplasia such as CHH, and microcephaly syndromes such as Nijmegen breakage syndrome. While the severity of a cellular defect is often variable, some of them may manifest as classical SCID with life-threatening infections and autoimmunity.
For an infant with congenital heart malformation, facial dysmorphism, and palatal defect, it is important to look for hypocalcemia and lymphopenia, which suggest 22q11.2 deletion syndrome or CHARGE syndrome.63 Further immunological investigations, including for lymphocyte subset and Ig levels, should be initiated. As blood transfusion and platelet support are often required when these infants undergo cardiac operation, the blood bank should be notified to provide irradiated, CMV-negative blood products if lymphopenia is apparent.
Table 50-4 summarizes the various forms of syndromic immunodeficiencies that can be recognized at birth because of body dysmorphisms and susceptibility to infections. In some forms of syndromic immunodeficiencies, immune aberrations progress with age and susceptibility to infections occurs later in childhood, such as ataxia telangiectasia. Review of the full spectrum of syndromic PID is beyond the scope of this chapter, and readers can refer to a few excellent reviews by Ming et al64,65 and Kersseboom et al.66
Table 50-4 Somatic and Immunological Features of Syndromic Primary Immunodeficiencies
Cytopenia
A number of PIDs are associated with cytopenia caused by immune-mediated destruction or bone marrow failure.67 As described in a previous section, patients with leaky SCID may develop immune cytopenia. Infants with WAS commonly present with petechial rash and eczematous skin lesions, and in severe cases, major bleeding such as intracranial hemorrhage may result from profound thrombocytopenia. Autoimmune hemolytic anemia and neutropenia may coexist.68 Autoimmune cytopenia affects more than 70% of patients with ALPS, and hypersplenism may cause consumptive thrombocytopenia, which is often severe and refractory to treatment.44,45 In reticular dysgenesis, arrest in myeloid differentiation leads to neutropenia,16 while patients with dyskeratosis congenita develop aplastic anemia.69 Fever unresponsive to antimicrobials, hepatosplenomegaly, and cytopenia are cardinal signs of HLH, and further laboratory investigations are required to fulfill the diagnostic criteria.42 Infants with cytopenia are often brought to the attention of hematologists, and the constellation of infectious and autoimmune complications should prompt the consideration of respective PID syndromes.
Family history
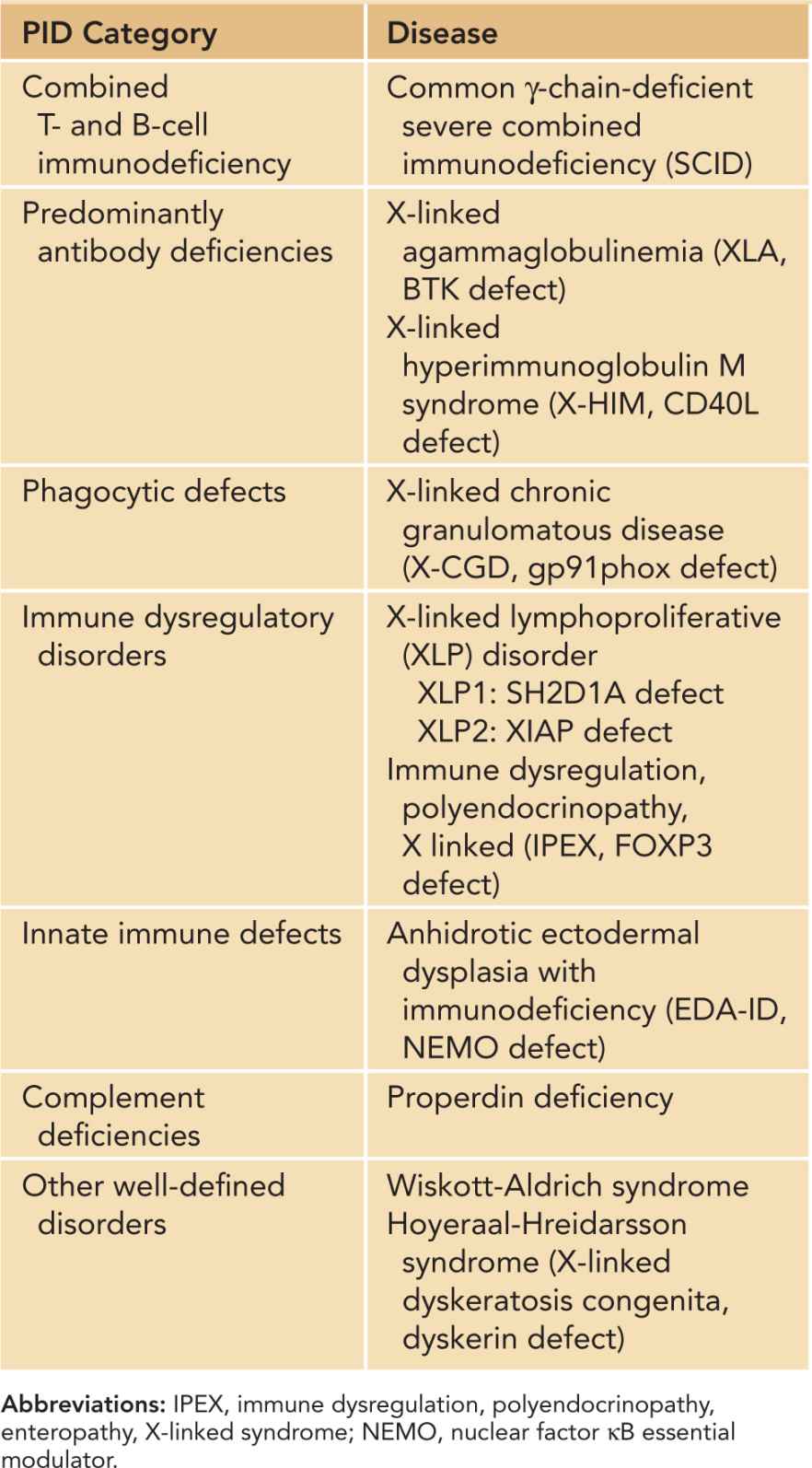
Family history of recurrent infections, autoimmune disease, malignancy, and unexplained infant deaths should be elicited to determine the pattern of inheritance. Presence of affected male relatives on the maternal side is particularly relevant, as X-linked SCID accounts for more than 50% of all forms of SCID.70 Other X-linked PIDs are listed in Table 50-5.71 The risk of AR disorders is increased in consanguineous families, which are prevalent in the Middle East; northern and sub-Saharan African; and western, central, and southern Asia. Some PIDs are also more prevalent in certain ethnic populations due to founder mutations, such as Artemis-deficient SCID in Athabascan-speaking native Americans72 and CHH in the Amish population.73
Table 50-5 X-Linked Primary Immunodeficiencies (PIDs)
INVESTIGATIONS
Diagnostic investigations of an infant with suspected PID follow a stepwise approach, from initial screening, immunophenotyping, functional characterization, to molecular diagnosis.74 It should be noted that age-specific reference range, ideally locally derived, has to be used.
Complete Blood Cell Count
Complete blood cell count provides useful information as an initial evaluation. It has to be emphasized that infants have high levels of total lymphocytes, and an age-specific reference range should be used. The reference interval for absolute lymphocyte count (ALC) is 3.4–7.6 × 109/L for healthy newborns, and T cells normally make up 70% of circulating lymphocytes.74 An infant with an ALC less than 3.0 × 109/L is lymphopenic, and immediate immunologic evaluation is warranted if confirmed on a repeat test. It should be noted that ALC can be normal or increased in Omenn syndrome and maternal T-cell engraftment.75
Chest X-ray
The normal thymus is visualized on the chest x-ray as a prominent soft-tissue density in the anterosuperior mediastinum up to the age of 3 years.76 Narrowing of the anterior mediastinum is suggestive of thymic hypoplasia. The chest x-ray of an infant with severe T-cell deficiency often shows features of interstitial pneumonia commonly caused by respiratory viruses or PCP. Cupping and flaring of the costochondral junction of the ribs characteristic of ADA deficiency may be appreciated.48
Lymphocyte Subset
The total number and percentage of T cells (CD3+) and their major subsets (CD4+ and CD8+), B cells (CD19+), and NK cells (CD16+/CD56+) in the peripheral blood are determined by flow cytometry. This provides information on the degree of numerical deficiency and immunophenotype that provides clues to the underlying molecular diagnosis.75,77 T−B+ phenotype suggests defects in the signaling pathway mediated by cytokines belonging to the γc family, while T−B− phenotype indicates V(D)J recombination defects. Lymphopenia is more profound in ADA SCID and reticular dysgenesis and may exhibit near absence of all T, B, and NK cells. Selective absence of CD4+ T cells suggests MHC class II deficiency, while ZAP70 deficiency should be considered when CD8+ T cells are absent.75,78 IPEX is diagnosed by the absence of FOXP3-expressing CD4+CD25+ T cells. CD3+TCRαβ+CD4−CD8− double-negative T cells are increased in ALPS.75
Antibody Assays
The level of serum IgG in infants under the age of 6 months reflects passive transfer of maternal IgG across the placenta. Therefore, infants with SCID have apparently normal serum IgG despite their intrinsic failure of immunoglobulin production. IgA and IgM levels are normally low at birth. Immune dysregulation may lead to aberrant production of IgM. Serum IgE is elevated in Omenn syndrome, hyper-IgE syndrome, and IPEX.79
Older infants who have received routine vaccination can be tested for functional antibodies toward vaccine antigens, such as tetanus, diphtheria, and pneumococcus. These are absent in severe T-cell deficiency due to the lack of T-cell help for specific antibody response and primary B-cell deficiency.
These tests are the basic initial investigations that should be performed for infants suspected to have T-cell deficiencies. Pediatric immunologists should be consulted for clinical evaluation and specialized investigations to further delineate the immunophenotype and confirm the molecular etiology.
Lymphocyte Function
The ability of lymphocytes to proliferate in response to mitogens (phytohemagglutinin [PHA], concanavalin A [ConA], pokeweed mitogen [PWM]); antigens to which the individual is previously sensitized (eg, candida, purified protein derivative [PPD]); or monoclonal antibodies specific to T-cell surface antigens mediating signal transduction (eg, anti-CD3, anti-CD28) is evaluated by lymphocyte proliferation assay. Peripheral blood mononuclear cells are cultured with standard concentrations of stimulants, and incorporation of radiolabeled DNA precursor (tritiated thymidine) into the proliferating lymphocytes is evaluated at the end of the incubation period by a scintillation counter.75,78 Lymphocyte proliferation is typically absent in SCID and other severe T-cell deficiencies.
Analysis of T-Cell Origin and Clonality
Omenn syndrome and maternal engraftment can be distinguished by studying the origin of the T cells. Maternal engraftment can be confirmed by X/Y fluorescence in situ hybridization (FISH) if the patient is a boy, HLA typing, or analysis of short-tandem repeats. Maternal T cells can be detected in 50% of B−SCID and 80% of B+SCID. Most of them have a mature T-cell phenotype (CD45RO+).31,34
Clonality of T cells can be studied by analyzing the distribution of different TCR β-chains (Vβ) by flow cytometry or quantitative polymerase chain reaction (PCR). A restricted TCR repertoire can be observed in B−SCID with residual V(D)J recombination capacity.80 These oligoclonal T cells are activated and express activation markers such as HLA-DR and CD25.
Evaluation of Thymic Output
The majority of T cells (>90%) in newborns are naïve cells that are able to respond to neoantigens. They bear the surface marker CD45RA. After antigen encounter, a proportion of T cells differentiates into memory T cells that express CD45RO. The generation of naïve CD4+ and CD8+ T cells is a measurement of thymic output.81
Naïve T cells that exit the thymus contain circular DNA fragments generated during V(D)J recombination, known as T-cell receptor excision circles (TRECs). TRECs can be quantified by real-time quantitative PCR and is a reliable surrogate marker for enumerating recent thymic T-cell emigrants. Naïve T cells and TRECs are diminished in severe T-cell deficiency regardless of underlying molecular etiology. The assessment of TRECs together with TCR clonal distribution provides valuable information to the severity of T-cell defect and peripheral T-cell homeostasis.82 TRECs assay is also applied in NBS for SCID.83,84
Other Specialized Tests
Diagnosis of ADA and PNP deficiency can be established by measuring enzyme activity and concentrations of dATP (deoxyadenosine triphosphate) or dGTP (deoxyguanosine triphosphate) in red blood cells, respectively. Urine for deoxyadenosine is also useful for diagnosis of ADA SCID.85 It should be noted that these tests become invalid if the patient has received a red cell transfusion.
Patients with B−SCID may have DNA repair defects (Artemis, ligase IV, DNA-PKcs, Cernunnos deficiencies) leading to radiosensitivity. This can be evaluated by radiosensitivity assay and the capacity of DSB repair induced by γ-irradiation of cultured skin fibroblasts obtained by skin biopsy.86 These tests are performed in specialized laboratories and may not be widely available.
Specific laboratory tests are needed to establish the diagnoses of HLH and ALPS according to consensus diagnostic criteria, as summarized in Table 50-687 and Table 50-7,88 respectively.
Table 50-6 Diagnostic Criteria for Hemophagocytic Lymphohistiocytosis (HLH)



