The Biliary Tract
MORPHOGENESIS OF THE BILIARY TRACT
Colin D. Rudolph
In the human embryo the first anlage of the bile ducts and liver is the hepatic diverticulum from the proximal gastrointestinal tract, as described in Chapter 418. The caudal part of this bud, known as the pars cystica, grows in length and forms the gallbladder, cystic duct and common bile duct. At about the eighth week of gestation, the hepatic precursor cells that lie adjacent to the hilar portal vein vessels form a sleeve-like double layer of cells that extends toward the periphery along the smaller intrahepatic portal vein branches. These hepatoblasts strongly expresses biliary specific cytokeratins and can be considered biliary precursor cells, that then form a continuous single-layered ring around the portal mesenchyme, known as the ductal plate.1 Beginning at 12 weeks of gestation and extending into the postnatal period, the ductal plate undergoes progressive remodeling. As new ductules form they are incorporated into the periportal mesenchyme that surrounds the portal vein branches. Thus, during successive periods of fetal life, ductal plate remodeling leads to the formation of the intrahepatic biliary tree. The largest ducts are formed first, followed by segmental, interlobular, and, finally, the smallest bile ductules. Arrest or derangement in remodeling leads to the persistence of primitive bile duct configurations termed ductal plate malformations. The occurrence of ductal plate malformations at different generations of the developing biliary tree gives rise to different clinicopathologic entities, such as congenital hepatic fibrosis and Caroli syndrome.
CONGENITAL GALLBLADDER ABNORMALITIES
A variety of structural abnormalities of the gallbladder have been described. Congenital absence of the gallbladder occurs in one of 7500 to 10,000 people. Failed development of the pars cystica is the likely etiology. As an isolated abnormality this is often of little clinical significance, although symptoms of abdominal pain, nausea and fatty food intolerance may develop because calculi form in the ductal system. In these patients gallbladder agenesis is frequently misinterpreted as cholecystitis with cystic duct obstruction.2 In addition to extrahepatic biliary atresia, which may accompany agenesis of the gallbladder, other associations with an absent gallbladder include imperforate anus, genitourinary anomalies, anencephaly, bicuspid aortic valve, and cerebral aneurysms. Hypoplasia of the gallbladder has also been described in association with neonatal diabetes, hypoplastic pancreas, and intestinal atresia.3 There is also an association with trisomy 18. The incidence of double gallbladder is 0.1 to 0.75 per 1000. The two cystic ducts may converge into a single duct, forming a Y-shaped structure. The accessory gallbladder may lie under the left lobe of the liver, draining into the left hepatic duct. A “floating gallbladder” is an anatomic variant observed in up to 5% of individuals. The gallbladder lacks a peritoneal coat or supporting membrane, making the pendulous gallbladder susceptible to torsion. This presents clinically as acute, severe right upper quadrant pain with nausea and vomiting. Often symptoms follow rapid movements that generate centrifugal forces causing gallbladder torsion and volvulus. Symptoms are consistent with an acute cholecystitis requiring operative intervention.4
DUCTAL PLATE MALFORMATIONS
Gregorz Telega and Colin Rudolph
CONGENITAL HEPATIC FIBROSIS
Congenital hepatic fibrosis (CHF) is the most common ductal plate malformation. It is most commonly associated with autosomal-recessive polycystic kidney disease (ARPKD) and occurs with an incidence of one in 6000 to 40,000 births (see Chapter 470).5 A large number of mutations in the PKHD1 gene associated with ARPKD and CHF have been identified but the pathogenicity of the defects has as yet not predicted the clinical phenotype.6 Congenital hepatic fibrosis may also be frequently associated with von Myenburg complexes (bile duct microhamartomas), and autosomal-dominant polycystic kidney disease. Less frequently it can be associated with renal dysplasia, nephronophthisis, asphyxiating thoracic dystrophy (Jeune syndrome), Bardet-Biedl syndromes and congenital disorder of glycosylation, type 1b (phosphomannose isomerase deficiency).
 CLINICAL FEATURES
CLINICAL FEATURES
Hematemesis or melena is the presenting sign in 30 to 70% of patients. It may occur as early as 1 year but more typically at 5 to 13 years of age. Gastrointestinal bleeding is a consequence of portal hypertension caused by fibrosis of the liver. Physical examination reveals abdominal distention with a firm, enlarged liver, often with a prominent left lobe, and splenomegaly. Many patients have enlarged palpable kidneys. In most patients the biochemical parameters of hepatic synthetic function are normal, and there is occasionally a mild elevation of aminotransferase values. Irregular bile ducts resulting from the ductal plate malformation present a risk of ascending cholangitis, which significantly contributes to morbidity and mortality. Abnormal portal flow can cause clot in the portal vein and result in development of the cavernous transformation of the portal vein. Patients with cavernous transformation of the portal vein appear to be at higher risk for more severe portal hypertension and complications of variceal bleeding.7
Due to association with ARPKD many patients with CHF will have arterial hypertension or uremia. In neonates or infants who present with CHF-ARPKD complications of the renal disease dominate clinical picture, whereas those who present later in childhood or in adulthood have a predominance of hepatic complications.
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Diagnosis is confirmed by liver biopsy. The hepatic lesions are fairly uniform and rarely give rise to macroscopically visible cysts. Microscopically characteristic dysmorphic cystic structures lined with columnar biliary epithelium in the portal zones, surrounded by dense fibrous deposits are found. These correspond to incompletely remodeled ductal plates. The portal tracts often lack normal interlobular ducts in the center. The abnormal dilated branching bile duct structures are in continuity with the rest of the biliary system.
Treatment is similar to that of other chronic liver disorders and includes nutritional management, including administration of fat soluble vitamins, and management of complications of cirrhosis if required. Variceal bleeding is treated with either variceal band ligation or sclerotherapy. Beta blockers, variceal band ligation, sclero-therapy or portosystemic shunting can be used as a prophylaxis of the variceal bleeding. Aggressive antibiotic therapy should be administered for cholangitis. In patients with chronic cholangitis and/or progressive hepatic dysfunction, liver transplantation may be the best treatment option. In isolated congenital hepatic fibrosis, the prognosis is good, given the excellent preservation of parenchymal and renal function in most patients.
CAROLI SYNDROME
Caroli disease is a congenital ductal plate disorder associated with congenital dilatation of the larger, segmental intrahepatic bile ducts. When this lesion is combined with the changes of congenital hepatic fibrosis, as is typically the case, the disorder is termed Caroli syndrome.8 Inheritance patterns are consistent with an autosomal-recessive disease. Both occur with an incidence of less than 1 per 100,000. A genetic cause is likely. Mutations in the APRKD gene, PKHD1, have been found in some but not all patients with Caroli disease.
 CLINICAL FEATURES
CLINICAL FEATURES
Caroli disease may present at any age but is most frequently recognized in adolescents or young adults with recurrent bouts of cholangitis and abscesses caused by bile stasis and gallstone formation within cysts, including fever, pruritus, jaundice, a tender liver, and modest elevations in serum bilirubin, alkaline phosphatase, and aminotransferase. Patients with Caroli syndrome may also have hepatic fibrosis, as seen in congenital hepatic fibrosis. Both Caroli disease and Caroli syndrome are associated with a 100-fold increased risk of cholangiocarcinoma compared to the general population.
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Liver biopsy may reveal the changes of congenital hepatic fibrosis. Diagnosis traditionally has been made by ultrasonography, computed tomography, percutaneous transhepatic cholangiography, or ERCP but MR cholangiography is emerging as the method of choice.9 Saccular and cystic dilation of the intrahepatic bile ducts and enlargement of the major intra- and extrahepatic biliary passages are evident (Fig. 427-1). Ursodeoxycholic acid therapy may enhance biliary flow and decrease the frequency of cholangitis episodes.10 Cholangitis should be treated with broad spectrum antibiotics, including anaerobic coverage. Partial hepatectomy may be helpful when disease is primarily confined to a single lobe.11 These measures are only partially successful in most patients with continuing problems of sepsis, cholangiocarcinoma, and amyloidosis. Liver transplantation is reported to be curative in patients with recurrent cholangitis, sepsis, or cholangiocarcinoma.12
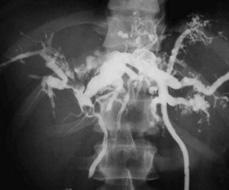
FIGURE 427-1. Caroli disease. Percutaneous trans-catheter cholangiography shows dilated bile ducts with stones and abscess. (From Lendoire J, et al: Bile duct cyst type V (Caroli’s disease): surgical strategy and results, HPB 2007;9(4):281-284.)
CHOLEDOCHAL CYSTS
The incidence of choledochal cysts has been estimated between one in 13,000 and one in 2,000,000 live births, and they are found in girls four times more frequently than in boys. Choledochal cysts are more prevalent in Asians, specifically the Japanese. The etiology of cyst formation is unclear, although there is growing evidence that the dilatation results from an anomalous junction of the common bile duct and the pancreatic duct, resulting in a common channel that is as long as 3.5 cm, versus a normal of 5 mm. This long common channel may allow for the reflux of pancreatic proteases into the extrahepatic biliary tree, resulting in cholangitis and stenosis.13 This hypothesis is supported by the measurement of high levels of amylase within the cysts, but the documentation of prenatal choledochal cysts suggests alternative abnormalities in the process of morphogenesis.
 CLINICAL FEATURES
CLINICAL FEATURES
Jaundice is the most common presentation of choledochal cysts in infants, being diagnosed in about 2% of infants presenting with cholestasis. In older children and adults abdominal pain is as common a presenting symptom as jaundice.14,15 Eighty percent of patients are female. Most patients present within the first decade of life, with the diagnosis made in 38% in the first year of life, 34% between ages 1 and 6 years, and 28% thereafter. The classic triad of abdominal pain, jaundice, and palpable right upper quadrant mass occurs in some patients, but in recent years the diagnosis is more commonly made during abdominal imaging for non-specific pain symptoms.16 Other common symptoms are fever, nausea, and vomiting, with or without associated pancreatitis.
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Children with choledochal cysts may have abnormal aminotransferase, bilirubin, and pancreatic enzyme values. Early treatment is required to prevent hepatic complications including fibrosis.16 Ultrasound is the most valuable screening test, as it can demonstrate both intrahepatic and extrahepatic dilatation of the biliary tree. Radionuclide scanning may demonstrate the accumulation of tracer within the cyst. Endoscopic retrograde cholangiopancreatography provides better delineation of the biliary anatomy. On occasion, impacted biliary calculi within the distal biliary tree are removed, resulting in resolution of the biliary dilatation. The location of the cyst allows classification into one of five anatomic types, as described by Todani (Fig. 427-2).18 The overwhelming majority of choledochal cysts are of type I, with diffuse enlargement of the common bile duct. Complications consist of perforations, liver abscesses, stone formation, secondary biliary cirrhosis, pancreatitis, amyloidosis and carcinoma of the biliary tree. Co-existing biliary anomalies including double common bile duct, double gallbladder, absent gallbladder, annular pancreas, biliary atreisa or stenosis are found in up to 25% of cases. Choledochal cysts vary in size with some of the larger ones containing 5-10 liters of bile.
The most worrisome complication of choledochal cyst is the high incidence of associated malignancy. In Japan, and incidence of 2.5 to 17.5% is reported. The majority of tumors are adenocarcinomas, detected at a mean age of 35 years.18 The cause of the malignant transformation is unclear but may relate to the chronic reflux of pancreatic proteases as well as the mutagenic potential of secondary bile acids in a stagnant environment. This has led to the recommendation that cysts be completely excised with concurrent removal of the gallbladder because of the risk of neoplasia. The approach to biliary reconstruction varies depending on the cyst type, anatomy, and surgical preference; the most common procedures are hepaticoduodenostomy, hepaticojejunostomy, or jejunal interposition.
BILE DUCT PAUCITY SYNDROMES
Vincent F. Biank and Colin D. Rudolph
Bile duct paucity syndromes are divided into those that are syndromatic or nonsyndromatic. Nonsyndromatic bile duct paucity is relatively rare compared to the syndromic form known as Alagille syndrome. Paucity of the intrahepatic bile ducts is a histologic finding defined as a ratio of interlobular ducts to portal tracts less than 0.9. However, since the remodeling of the intrahepatic bile ducts in the most peripheral (smallest) portal tracts is not complete until approximately 6 weeks of age, defining the “normal” bile duct-to-portal tract ratio to estimate the degree of ductopenia in the liver is problematic in the premature infant, where a ratio lower than 0.9 may be normal.19
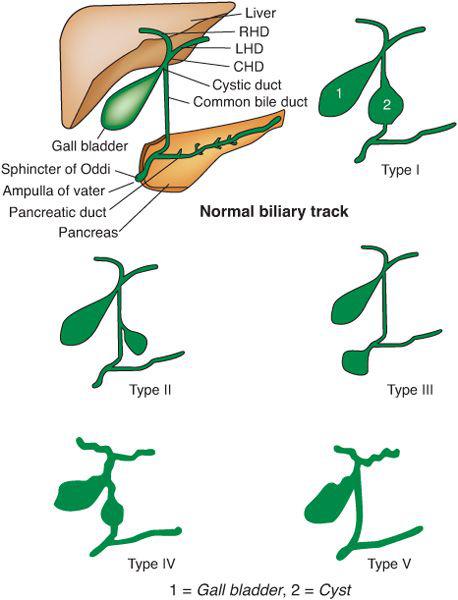
FIGURE 427-2. 5 types of Choledochal cysts. Type I: dilatation of extrahepatic biliary duct; Type II: Cyst from common bile duct (CBD); Type III: choledochocele or dilatation of distal part of CBD, type IV: dilatation of both extrahepatic and intrahepatic duct; Type V: Caroli disease, Dilatation of intrahepatic duct only. CHD: common hepatic duct, LHD: right hepatic duct and RHD: right hepatic duct. From: http://en.wikipedia.org/wiki/File:Choledochal_cysts.svg
ALAGILLE SYNDROME
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
Alagille syndrome is a multisystem, inherited disorder with highly variable clinical features even within affected families. The incidence is about 1 in 70,000 live births. Mutations of the JAG1 gene, that encodes protein ligands for NOTCH1, are detected in over 88% of individuals with Alagille syndrome.20 Fluorescence in situ hybridization (FISH) detects a microdeletion of 20p12, including the entire JAG1 gene, in approximately 7% of affected individuals. Mutations in NOTCH2 are observed in fewer than 1% of individuals with Alagille syndrome. The NOTCH proteins play a role in determining cell fate during differentiation, especially in tissues where epithelial-mesenchymal interactions are important. The disorder is inherited in an autosomal dominant manner. Approximately 30%-50% of individuals have an inherited mutation and about 50%-70% have a de novo mutation. The offspring of an individual with Alagille syndrome have a 50% chance of having Alagille syndrome. Prenatal testing is possible if the JAG1 disease-causing mutation or a deletion detected by FISH is identified in an affected family member. However, although genetic testing can determine whether or not the fetus has inherited the JAG1 disease-causing mutation or deletion, it cannot predict the occurrence or severity of clinical manifestations.
 CLINICAL FEATURES
CLINICAL FEATURES
Alagille syndrome was initially described as an association of congenital heart disease and neonatal cholestasis. The most frequent cardiovascular findings are peripheral pulmonary artery stenosis but more severe hypoplasia of the pulmonary artery branches may occur. Complex congenital heart disease such as tetralogy of Fallot has been found. Other common clinical features include skeletal abnormalities and a typical facies. The characteristic facies in the infant or child has the shape of an inverted triangle. The forehead is broad, and eyes are deep-set with mild hypertelorism; the nose is small and straight, and the chin is small and pointed. The facies may not be evident in the first months of life. The classic childhood appearance differs from its adult form, which may have somewhat coarse facial features, often with a long face, deep-set eyes, and prominent forehead.
Butterfly vertebrae, from failure of the anterior arches of the vertebral body to fuse, are most commonly detected in the thoracic spine. Other vertebral abnormalities have been described such as abnormalities of the interpedicular distance in the lumbar spine and spina bifida occulta. Very short distal phalanges and a short ulna have also been reported. Eye findings include posterior embryotoxon, optic disc drusen, retinal abnormalities (including abnormal pigmentation but not functional retinal degeneration), strabismus, ectopic pupil, and hypotrophic optic discs.
Renal disease associated with Alagille syndrome is highly variable. Structural abnormalities include symmetrically small kidneys or congenital single kidney. Several reports document renal cystic disease associated with Alagille syndrome. Histologic examination of kidneys in Alagille syndrome has revealed a membranous nephropathy in some cases, but the most frequent finding is lipid accumulation in the kidney (mesangiolipidosis). Nonspecific changes include azotemia, defects in concentrating urine, and nephrolithiasis.
Other abnormalities include small birth size and/or poor growth, delayed puberty or hypogonadism, abnormal cry/voice (“high-pitched”), and mental retardation, learning difficulties, or antisocial behavior. Associated vascular abnormalities have been noted including decreased intrahepatic portal vein radicals, moyamoya disease, coarctation of the aorta, and anomalies of other large arterial vessels. Neurologic abnormalities, especially peripheral neuropathies, described in early reports probably were not part of the syndrome itself but due instead to vitamin E deficiency from severe chronic cholestasis. Hypothyroidism and pancreatic insufficiency have also been observed in association with Alagille syndrome.
 DIAGNOSIS
DIAGNOSIS
In most cases a clinical diagnosis is made based upon findings of bile duct paucity on a liver biopsy that is performed as part of the evaluation of neonatal cholestasis. Although considered to be the most important and constant feature of Alagille syndrome, bile duct paucity is present in only 90% of infants with the disorder. A normal ratio of portal tracts to bile ducts, bile duct proliferation, or a picture suggestive of neonatal hepatitis may be observed. Surgical biopsy is usually required to examine at least 20 portal tracts, the recommended sample number for definitive diagnosis. However, many experts believe that visualization of as few as five portal tracts in a percutaneous biopsy may be sufficient for diagnosis in the appropriate clinical setting.22 In addition to bile duct paucity, three other features including cholestasis, cardiac defects, skeletal abnormalities, characteristic facies or ophthalmologic abnormalities are required for diagnosis. Evaluation otherwise includes an echocardiogram to evaluate for cardiac anomalies and peripheral pulmonic stenosis, AP and lateral chest radiographs to allow evaluation for the presence of butterfly vertebrae, ophthalmologic examination to identify anterior chamber involvement, renal ultrasound and renal function testing to identify renal complications, and screening for early identification of any developmental delays. Confirmation of the diagnosis by genetic testing for JAG1 mutations is desirable. Given the medical problems of this condition and their variability, it is appropriate to assess first-degree relatives for manifestations of the disorder. If a JAG1 mutation has been identified in a proband, at-risk relatives can be evaluated using genetic testing. If no JAG1 mutation has been identified, at-risk relatives are best assessed with measurement of liver enzymes, cardiac examination, eye examination, skeletal x-rays, and evaluation of facial features.
 TREATMENT AND OUTCOMES
TREATMENT AND OUTCOMES
Alagille syndrome has a variable course. Patients with Alagille’s syndrome should not undergo Kasai portoenterostomy since it will not improve bile flow. Close monitoring of plasma concentration of fat-soluble vitamins, nutritional optimization, and therapy with vitamins A, D, E, and K prevent fat soluble vitamin deficiency. Zinc deficiency may also occur and require replacement therapy. The cholestasis usually improves or resolves over the first year of life. If this occurs the patients generally do not develop cirrhosis. In about 15% of Alagille syndrome patients the liver disease progresses to cirrhosis with eventual liver failure that requires liver transplantation. Comparatively minor head trauma may cause significant intracranial bleeding in infants and toddlers with Alagille syndrome; mortality is significant, even in the absence of coagulopathy.
Pruritus can be very severe and disabling. Pruritus can be treated with rifampin or naltrexone.23 Hyperlipidemia and hypercholesterolemia can be very problematic with resultant xanthomas. The hyperlipidemia has been treated with cholestyramine but efficacy of such therapy is uncertain. Xanthomas have been successfully treated with choloretic agents (ursodeoxycholic acid) cholestyramine or partial external biliary diversion. Liver transplantation for end-stage liver disease has an 80.4% five-year survival rate, and results in improved liver function and some catch-up growth in 90% of affected individuals.24
Conservative estimates put overall mortality at 20 to 25% from cardiac disease, intercurrent infection, or progressive liver disease. Neurovascular accidents have also been reported with rates as high as 15% and account for up to a third of the mortality in Alagille syndrome.25 Renal failure is another potential long term complication from Alagille syndrome. Hepatic malignancy has been reported in both children and adults.
NON-SYNDROMATIC BILE DUCT PAUCITY
Paucity of the bile ducts has been described in association with a wide variety of conditions including Down syndrome, Turner syndrome, hypopituitarism, cystic fibrosis, alpha-1-anti-trypsin deficiency, congenital infections such as cytomegalovirus, rubella, and syphilis, hepatitis B, graft-versus-host disease, chronic hepatic allograft rejection, primary sclerosing cholangitis, Zellweger syndrome, and Ivemark syndrome. Treatment of the liver disease is similar to that of syndromatic bile duct paucity. Other manifestations of the associated disorders are treated accordingly.
EXTRAHEPATIC BILIARY ATRESIA
Vincent F. Biank and Colin D. Rudolph
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



{kind=link}