T-Cell Dysfunction
NEONATAL T-CELL FUNCTION
T cells are major effector cells of the adaptive immune response from influencing antibody production to maintaining tolerance. The defining marker of the T cell is the T-cell receptor (TCR), and 2 distinct T-cell subsets have been characterized based on the structure of the TCR: TCRα/β and TCRγδ. The specificity of the TCRα/β T-cell repertoire is established during fetal development of the thymus, which develops at week 6 of gestation. Immature thymic cells develop from fetal liver cells and, after birth, from hematopoetic stem cells (HSCS) in the bone marrow. T cells develop from common lymphoid progenitor cells derived from HSCs within the thymus and undergo stages of differentiation, expressing 1 or both of the cytodifferentiation (CD) antigens CD4 and CD8. T cells expressing the α/β TCR are divided into various subsets based on surface CD antigens and function. CD4+ T cells, or helper T cells (TH), make up approximately 70% of T cells in the peripheral blood. TH cells recognize foreign antigens processed and presented in major histocompatibility complex (MHC) class II molecules and can be divided into additional subsets. TH1 CD4+ T cells produce interferon gamma (IFN-γ) and IL-2, favor antibody class switching to immunoglobulin (Ig) G2, and mediate protection against intracellular pathogens. TH2 CD4+ T cells produce IL-4, IL-13, and IL-5, favor class switching to IgG1 and IgE, and participate in atopic diseases and immunity against parasites. More recently, additional subsets of CD4+ T cells have been described that have effector functions against extracellular microbes: TH9 cells produce the anti-inflammatory cytokine IL-10 and are involved in the host defense against nematodes, whereas TH17 cells secrete IL-17, IL-21, and IL-22 and promote protective immunity against extracellular bacteria and fungi at mucosal surfaces. CD8+ cytotoxic T lymphocytes (CTLs) recognize endogenous proteins bound to MHC class I molecules, such as viral and tumor antigens, and mediate cytotoxicity by lysing altered or nonself cells. A third class of T cells (typically CD4+) with immune suppressive function are denoted as regulatory T cells (Treg) and can be identified (albeit with some exception) by the expression of the transcription facto FOXP3 (forkhead box P3). Treg cells are produced directly from the thymus (natural Treg) or induced within the periphery (induced Treg) and play major roles in mediating chronic inflammation, allergic disease, and autoimmunity.1
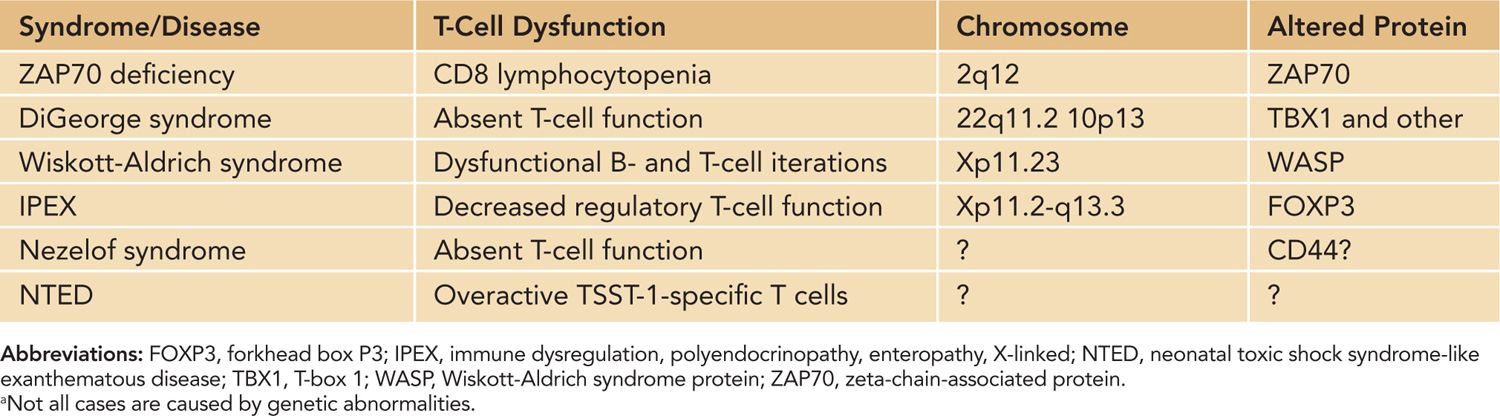
T-cell dysfunction in neonates has implications for a variety of responses and disease states. Infants are more susceptible to infectious agents, and according to the World Health Organization (WHO) estimates, approximately 2.5 million infants under the age of 1 die annually of infection. Infants are also less responsive to vaccination. In vaccinated infants, immunity wanes around 6–9 months after vaccination, and multiple booster shots are needed to maintain immunological memory.2 Historically, the function of neonatal T cells has been considered impaired, mainly attributed to immaturity of the neonatal immune response. Neonatal antigen-presenting cells produce lower amounts of IL-12, required for IFN-γ responses. The overall bias of the neonatal immune response toward TH2 leads to delayed production of proinflammatory cytokines and IL-2 and subsequent delayed effective CD8+ CTLs until several months after birth. The TH2 dominance during early life also influences the class of IgG antibodies, leading to a predominance of IgG1 and IgG3 and low production of IgG2. However, in certain instances, such as BCG vaccination, a proper TH1 response is mounted in neonates. Thus, baseline normal neonatal adaptive responses range from nonresponsiveness to adult-level T-cell responses (Table 112-1). Genetic defects in neonatal T-cell function tend to increase susceptibility of the already-vulnerable newborn to disease (Table 112-2). Because optimal B-cell function requires T-cell help, a T-cell defect typically results in a B-cell deficiency as well (considered combined immunodeficiencies, such as Wiskott-Alrich syndrome [WAS]). Newborns with primary immunodeficiencies most commonly present with increased cases of infection; however, diagnosing the disease with known characteristics prior to infection is the goal and will typically lead to a better outcome for the patient.
Table 112-1 Neonatal T-Cell Responses to Vaccines and Infectious Disease

Table 112-2 Inherited T-Cell Immunodeficiencies and Disease
CONGENITAL T-CELL IMMUNODEFICIENCIES
DiGeorge Syndrome
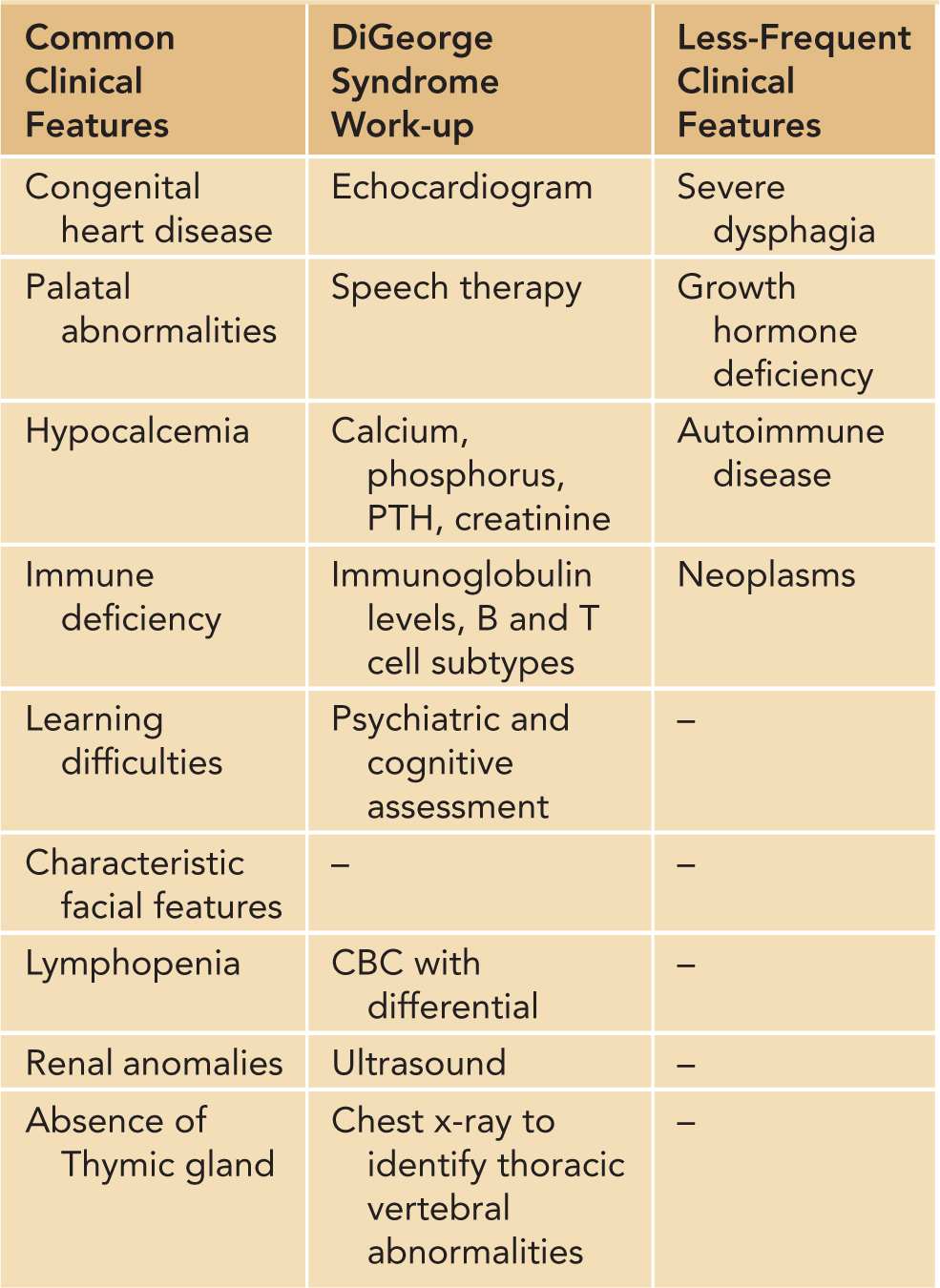
DiGeorge syndrome,3 or 22q11.2 deletion syndrome, is caused by the deletion affecting the long arm of chromosome 22. It is a genetic recessive immunodeficiency (in rare cases autosomal dominant) usually inherited from the mother. DiGeorge syndrome has occurred in both males and females and has an estimated prevalence of 1:4000. Some cases of DiGeorge and velocardiofacial syndromes have defects in other chromosomes, notably a deletion in chromosome region 10p14. There is considerable variability in the phenotype, and severity is based on the extent of the microdeletion. Individuals with DiGeorge syndrome may suffer from congenital heart defects, palatal abnormalities, hypocalcemia caused by hypoparathyroidism, distinctive craniofacial features, renal anomalies, and thymic hyplasia.4 Additional clinical findings associated with the disease and useful evaluations following diagnosis with DiGeorge/22q11.2 deletion syndrome are summarized in Table 112-3.
Table 112-3 Clinical Characteristics and Diagnosis of DiGeorge/22q11.2 Deletion Syndrome
T-cell immunodeficiency in DiGeorge syndrome is attributed to failure of development of the thymus, and patients may be mildly to severely lymphopenic based on the degree of thymic deficiency. Laboratory evaluation typically reveals normal-to-decreased numbers of T lymphocytes with absent-to-normal T-cell function and normal B-cell function.5 Serum immunoglobulin levels tend to be within normal limits. However, in the rare patient, B-lymphocyte function and antibody production may be abnormal as well. A defect in the delayed-type hypersensitivity is observed by the failure of affected patients to develop a positive skin test to candidin. Because of defects in cell-mediated immunity, patients are highly susceptible to opportunistic infections and to graft-vs-host disease (GVHD) from nonirradiated blood transfusions.
Diagnosis of individuals with DiGeorge syndrome is confirmed for the microdeletion on chromosome 22 via fluorescence in situ hybridization (FISH) chromosomal analysis.6 The 2 probes commercially available for 22q11.2 FISH analysis are TUPLE1 and N25. Fewer than 5% of individuals with clinical symptoms of the 22q11.2 deletion syndrome have normal routine cytogenetic studies and negative FISH testing. Fetuses at increased risk may be evaluated between 18 and 22 weeks’ gestation by high-resolution ultrasound examination for palatal and other associated anomalies, by echocardiography for cardiac anomalies, or by chromosomal analysis of fetal cells obtained through amniocentesis.
Treatment of the 22q11.2 deletion/DiGeorge syndrome is based on the severity of the disease and may require surgery for cardiac defects and vitamin D, calcium, or parathyroid hormone replacement to correct hypocalcemia and treat seizures. Most infants have a mild-to-moderate deficit in T-cell function that often improves with age and do not suffer from recurrent infections in adulthood. For patients with the complete form of the syndrome with absent T-cell immunity (<0.2%), thymic grafts have been used successfully. Human leukocyte antigen (HLA)-identical bone marrow transplants have been successful at developing T-cell function in these patients, but this treatment cannot correct the thymic defect. Of note, prior to giving live vaccines, T-cell numbers and function should be assessed, if not done earlier, to prevent vaccine-related side effects.
Wiskott-Aldrich Syndrome
Wiskott-Aldrich syndrome is an X-linked recessive immunodeficiency disease caused by mutations in the gene that produces the Wiskott-Aldrich syndrome protein (WASP). Various mutations in the WASP gene have been identified in patients with WAS, each affected family typically carrying its own characteristic mutation. If the mutation interferes completely with the ability to produce the WASP, the patient has the classic and more severe form of WAS. Alternatively, if the mutation only results in lower production of WASP, a milder form of the disorder may result. WAS is characterized by defective interactions between B and T cells, resulting in decreased cell-mediated immunity. Patients with classic WAS present with thrombocytopenia, eczema, and recurrent infection. Common infections are typically caused by pneumococcal bacteria, resulting in pneumonia, meningitis, or sepsis, but may involve all classes of microorganisms. The initial clinical manifestations of WAS may be present soon after birth or develop in the first year of life, with prolonged bleeding from the circumcision site, bloody diarrhea, or excessive bruising. Survival to adulthood is rare; infection, vasculitis, and bleeding are major causes of death.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


