Systemic Lupus Erythematosus, Overlap Connective Tissue Diseases, and Mixed Connective Tissue Disease
Earl D. Silverman
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
Systemic lupus erythematosus (SLE) represents the prototype of a pediatric autoimmune disease with the presence of autoantibodies as its hallmark. The incidence of SLE diagnosed prior to age 18 is approximately 10 to 20 new cases per 100,000 population per year with an overall prevalence of 1 to 2 cases per 1000 adolescents ages 12 to 18. Both the incidence and prevalence rates are higher in African Americans, Asians, Southeast Asians, and Hispanics. The female predominance (4–4.5:1) in pediatric patients is lower than in adults (9:1).1 The mean age at diagnosis is approximately 12 to 13 years, but presentation as young as age 3 or 4 is routinely reported.2 Presentation prior to age 1 is very rare and may manifest as congenital nephrotic syndrome.
Neonatal lupus erythematosus (NLE) must not be mistaken for early-onset SLE. NLE is a disease caused by the transplacental passage of maternal autoantibodies, and the fetus/neonate is an innocent bystander with a normal immune system that is not actively producing autoantibodies. However, some of these children may develop true SLE many years later. Early-onset pediatric SLE is a disease in which the child produces autoantibodies and the immune abnormalities are intrinsic to the child’s immune system.
 ETIOLOGY
ETIOLOGY
Although the triggering mechanisms are not fully defined, the production of autoantibodies is the hallmark of SLE. These autoantibodies are usually directed against histone, nonhistone, RNA-binding, cytoplasmic, and nuclear proteins. Antinuclear antibody (ANA) occurs in most, if not all, patients with SLE. Anti-DNA antibodies are present in approximately 50% to 60% of patients, while antibodies directed against the small nuclear ribonuclear proteins (anti-Sm and anti-70kDa RNP antibodies) occur in 40% to 50% of patients.3 Antibodies directed against small cytoplasmic ribonuclear proteins (anti-Ro and/or anti-La antibodies) occur in 30% to 40% of patients, anticardiolipin antibodies in 40% to 50% of patients, and rheumatoid factor 15% to 20% of patients. Antiribosomal P antibodies are present in approximately 25% to 30% of patients and may be associated with psychosis and depression.4,5
Antiphospholipid and anticardiolipin antibodies are detected in approximately 50% of patients.6 One specific antiphopholipid antibody worth noting is the lupus anticoagulant (LAC), an antibody that reacts with phospholipids in the reagent used in the partial thromboplastin time (PTT) determination. Patients with LAC do not bleed; instead, they have an increased incidence of deep vein thrombosis, thromboemboli, or, less commonly, arterial thrombosis. Antiphospholipid antibodies are associated with multiple neurologic manifestations including stroke, seizures, chorea, and other movement disorders; pseudotumor cerebri; and migraine headache; as well as with nonneurologic disorders such as thrombosis, thrombocytopenia, or recurrent abortion.6–9 Most, if not all, pediatric patients with evidence of venous thromboembolic disease will have evidence of LAC.
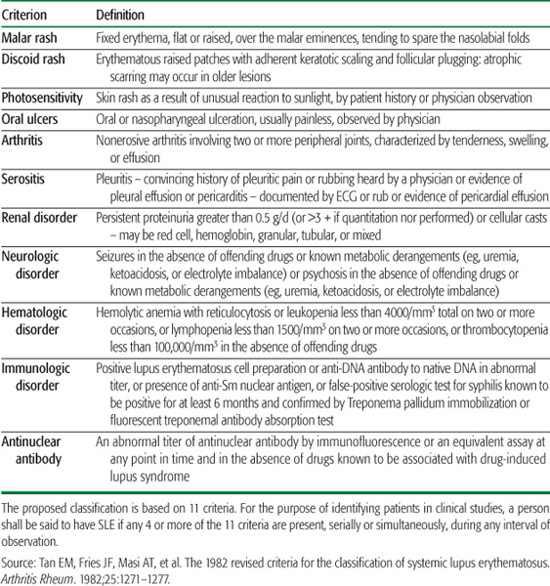
Table 204-1. 1982 Revised Criteria for Classification of Systemic Lupus Erythematosus
 CLINICAL PRESENTATIONS
CLINICAL PRESENTATIONS
In 1997 the American College of Rheumatology (ACR) revised the criteria for classification of SLE (Table 204-1).10 These criteria were not to be used to diagnosis and separate SLE patients from normal individuals but for classification of patients with an autoimmune into SLE as opposed to another autoimmune disease. Patients are classified as having definite SLE if they meet 4 out of 11 criteria, but most series report 10% to 15% of patients who have only 3 criteria.
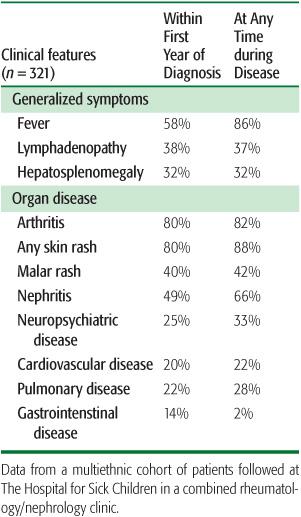
Arthritis, dermatitis, and nephritis are the most common manifestations, but any organ may be affected (Table 204-2). Systemic symptoms reflecting a generalized inflammatory process (fever, malaise, weight loss, and lethargy) are very common. In 80% to 90% of patients, a disease manifestation either occurs within the first year of diagnosis or it fails to arise. The exception is central nervous system (CNS) disease, in which up to 25% of patients will have their first episode of CNS involvement more than one year after diagnosis. Flares of SLE tend to be similar to the initial presentation.
Table 204-2. Frequencies of Clinical Features of Systemic Lupus Erythematosus in Children and Adolescents
Renal Disease
The incidence of renal involvement varies between 48% to 90% of patients.11-13 An extensive discussion of the diagnosis and management of renal complications of SLE is provided in Chapter 472.
Central Nervous System
Central nervous system (CNS) disease or neuropsychiatric (NP-SLE) involvement occurs in 20% to 40% of patients and is associated with significant morbidity and mortality.18-21 Both the central and/or the peripheral nervous systems may be involved with multiple syndromes and presentations. In 1999 the American College of Rheumatology developed a new nomenclature and case definition for neuropsychiatric SLE (eTable 204.1  ).22
).22
Psychiatric illnesses range from mood disorders to depression to frank organic brain syndrome. Neurocognitive testing detects impairments in cognitive function or learning difficulties in a high percentage of patients, but the true incidence is unknown.
Depression secondary to active disease must be differentiated from a secondary depression arising from environmental factors or from medication side effects. Overt psychosis or organic brain syndrome occurs in approximately 10% of all patients with SLE and may be attributed to endogenous CNS disease, metabolic imbalance, or infection, the latter sometimes precipitated by steroid therapy. In most patients with psychosis or organic brain syndrome, a lumbar puncture is indicated.
Seizures, seen in approximately 10% to 20% of patients, may be the presenting sign of more significant organic brain disease, the result of an infarction, or the sole manifestation of CNS involvement. Movement disorders encompass cerebellar ataxia, hemiballismus, tremor, parkinsonian-like movements, and chorea. SLE, or antiphospholipid antibody syndrome, is currently the most common cause of chorea in developed countries.23
Cranial nerve involvement is more common than peripheral neuropathy. Rarely, hemiparesis or transverse myelitis may occur. Although not studied in children, the incidence of autonomic dysfunction in adults is 40% to 50%, is usually mild, and may lead to changes in heart rate.
Headache occurs in up to 25% of patients with SLE. The typical headache responds to mild analgesia.24 However, a severe, unremitting headache, sometimes referred to as a lupus headache, usually reflects active disease or CNS vasculitis, or it may represent cerebral vein thrombosis. In all cases of unremitting headache, appropriate investigations must be performed to rule out cerebral vein thrombosis or infection.25 A more benign cause of headache is pseudotumor cerebri ascribable either to the underlying disease or to steroid medication. Migraine-like headaches are common and likely reflect active CNS SLE.
Examination and culture of cerebrospinal fluid (CSF) are performed to rule out the possibility of CSF infection or hemorrhage. An elevated CSF protein and/or CSF white blood cell count in the absence of infection is suggestive of cerebritis.20 Neuroradiologic investigation of the central nervous system (CT or MRI scan) may demonstrate specific structural lesions such as infarction, embolus, cerebral vein thrombosis, and subdural or intracranial hemorrhage, but these modalities are generally not helpful in measuring overall CNS disease activity.26 Levels of complement proteins and anti-DNA antibodies, which may correlate with disease activity at other sites, may be normal with CNS involvement.
The therapy of CNS disease varies with the manifestation. Active psychosis and/or organic brain syndrome are potentially life-threatening complications and should be treated aggressively with an immunosuppressive regimen that includes high-dose corticosteroids and azathioprine, MMF, or cyclophosphamide. Psychotropic drugs serve as adjunctive, but not primary, therapy.
Dermatologic Disease
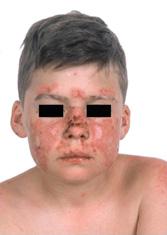
Skin involvement manifesting as malar rash, discoid rash, or photosensitivity occurs in 60% to 90% of patients with SLE.27 A rash in the malar area involving cheeks and nasolabial folds is quite specific; dermatomyositis is the only other disease in the differential diagnosis (Figs. 204-1 and 205-1). A discoid rash is rarer than a malar rash. Many but not all patients exhibit photosensitivity, and sun exposure may lead to a flare of skin and/or systemic disease. Sun-exposed areas should be protected with light clothing and a sunscreen with a high ultraviolet (UV) light protection rating against both UVA and UVB. The rash of subacute cutaneous SLE appears as an annular rash with a raised border and central sparing; it, too, has a photosensitive component and is often associated with anti-Ro and anti-La antibodies. Alopecia, listed in the original classification, occurs in 25% to 35% of patients. In pediatric patients, alopecia in the presence of a systemic autoimmune disease is quite specific for SLE. A vasculitic rash consisting of oral or nasal erosions or ulcers on the arms, legs, or ears may occur in up to 25% of patients and is often associated with systemic involvement (eFig. 204.1  ).
).
Musculoskeletal Involvement
As many as 90% of patients exhibit joint involvement, typically a polyarticular arthritis that affects both large and small joints; severe pain and significant morning stiffness occur in half the patients, whereas in the other 50% polyarthritis may produce few symptoms. Control of extra-articular sites of disease activity is frequently sufficient to treat the arthritis. An arthritic flare may herald a more generalized flare. Therapy with nonsteroidal anti-inflammatory agents and antimalarial agents may control arthritis as an isolated symptom, but low-dose corticosteroid therapy is frequently required.
Patients with SLE are at a high risk for the development of avascular necrosis (AVN) of many joints; this complication is likely secondary to a combination of the disease process, to antiphospholipid antibodies, and/or to the use of corticosteroids. AVN occurs in 5% to 10% of patients who present with acute pain, joint tenderness, and effusion. Septic arthritis and osteomyelitis must always also be considered if a fever is present.
Hematologic Involvement
Anemia, thrombocytopenia, and leukopenia occur in 50% to 75% of patients. Only a Coomb-positive hemolytic anemia satisfies the diagnostic criteria of the American College of Rheumatology, but both normochromic, normocytic anemia and microcytic, hypochromic anemia are more common in SLE. Thrombocytopenia is present in 30% to 45% and may precede the diagnosis of SLE. SLE should be considered in all children and adolescents with chronic thrombocytopenia.28-30 Leukopenia occurs in 20% to 40% of cases (lymphopenia and/or granulocytopenia). Pancytopenia may also occur, and when present one must consider the concomitant presence of a herpes family viral infection or macrophage activation syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


