Genetic Disorders of the Skin
Brook E. Tlougan and Amy S. Paller
SELECTED GENETIC DISORDERS OF THE SKIN
During the last decade, our understanding of the molecular bases for genetic disorders of the skin has expanded tremendously. Identifying the gene mutations that lead to phenotypic manifestations facilitates prenatal diagnosis using molecular techniques. For some disorders, this information has translated into early trials of gene therapy or the development of new pharmacologic therapy based on manipulation of gene product levels. 
THE ICHTHYOSES AND ICHTHYOSIFORM DISORDERS
Named for the Greek term meaning “fishlike scales,” this heterogeneous group of disorders is characterized by the predominant clinical feature of visible accumulation of scale. FIRST (Foundation for Ichthyosis and Related Skin Types) is a national support group for patients with the ichthyoses and other disorders with thickening of epidermis (http://www.scalyskin.org). During the past decade, the underlying molecular basis for many of the ichthyotic disorders has been discovered, and many can be diagnosed prenatally through molecular analysis of genomic DNA obtained by chorionic villus sampling or amniocentesis. In general, therapy for these disorders is similar and is based on disease severity and tolerance rather than the specific type. During the neonatal and early infantile period, however, therapy should be limited to the frequent application of bland emollients, since use of topical medications with keratolytic agents during the first 6 months of life is usually unnecessary and risks significant absorption of potentially toxic substances (eg, absorption of lactic acid, salicylic acid).
Scaling in the genetic forms is usually either present at birth or has its onset within the first few years of life. Rarely, nongenetic causes lead to ichthyosis in pediatric patients. Causes of acquired ichthyosis include hypothyroidism, chronic renal insufficiency, malignancy (particularly lymphoma), malabsorption syndromes, essential fatty acid deficiency, sarcoidosis, and certain drugs (particularly hypocholesterolemic agents).
Ichthyosis vulgaris is the most common form of the ichthyoses.1,2 It is now known to result from mutations in the gene encoding filaggrin, a gene also recently implicated in atopic dermatitis. Approximately 1 in 20 Northern European individuals carry a mutation on 1 allele and have a mild form; homozygotes have a much more severe form. Thus, the disorder is not autosomal dominant, as was once thought, but is semidominant. Regardless of severity, the onset of scaling in ichthyosis vulgaris tends to be after 3 months of age and may become more prominent later during childhood or at puberty. Fine, white scales, often without much erythema, predominate on the exterior surfaces of the extremities, especially the legs (Fig. 360-1). There is an increased prominence of palmoplantar markings (hyperlinear palms) owing to mild to moderate thickening of the palms and soles. The majority of patients with ichthyosis vulgaris show a reduced granular layer of skin with decreased conversion of profilaggrin to filaggrin, the major protein in keratohyalin granules. The risk of atopic dermatitis is increased in patients with ichthyosis vulgaris, suggesting an important role of the defective epidermal barrier in the development of the dermatitis. 
Recessive X-linked ichthyosis occurs in 1 of 2000 to 6000 boys. Scaling is often more pronounced than in ichthyosis vulgaris, and scales tend to be larger and darker; the trunk is usually involved, but palms and soles are unaffected.1,2 The antecubital and popliteal flexures are usually spared, whereas the neck and periauricular areas are affected. The disease is caused by the absence of the microsomal enzyme steroid sulfatase (arylsulfatase C). Because of the deficiency of fetal placental steroid sulfatase, the first clue to diagnosis may be failure to initiate labor or of having labor progress in the pregnant mother. Abnormalities of the genitalia, particularly undescended testes, have been described in approximately 10% of patients. Minute, asymptomatic corneal opacities are present in half of adult patients. Diagnosis can be confirmed by measurement of enzyme activity in fibroblasts, leukocytes, amniocytes, or scales or by measurement of substrate (cholesterol sulfate) accumulation in scales or blood. The majority of cases result from deletion of the gene, which can be detected by fluorescent in situ hybridization (FISH) analysis. Approximately 10% of affected individuals have contiguous gene deletion with associated hypogonadism, anosmia, and mental retardation.
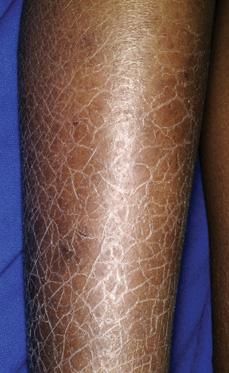
FIGURE 360-1. The scaling of patients with ichthyosis vulgaris is most severe on the lower extremities and during cold months. The palms and soles tend to be thickened as well in this semidominant common skin disorder. (Source: Courtesy of Amy Paller, MD.)
Epidermolytic hyperkeratosis, or bullous congenital ichthyosiform erythroderma, is an autosomal dominant trait in which large areas of denuded skin are typically present at birth, often suggesting a mechanobullous disease rather than a form of ichthyosis. The disease may be localized and mild or generalized and severe. A milder form, often with more superficial blistering and thinner scaling (ichthyosis bullosa of Siemens), results from mutations in keratin 2e, encoding keratin intermediate filaments expressed in the upper half of the epidermis. A more severe form with thicker scaling (bullous congenital ichthyosiform erythroderma of Brocq) is usually caused by mutations in either the keratin 1 or keratin 10 gene, which is expressed in the immediate suprabasal areas.  By infancy, scaling becomes more conspicuous. Blistering occurs less frequently with advancing age, is often focal, and may be caused by secondary staphylococcal infection. Although the disorder is generalized, scaling is particularly verrucous in intertriginous areas and overlying joints. The degree of associated erythroderma and palmoplantar keratoderma is variable. Treatment of this disorder with keratolytics or retinoids is often complicated by the propensity of these agents to increase skin fragility. The mosaic form of epidermolytic hyperkeratosis presents as epidermal nevi with linear streaks of thickening of skin, often with increased pigmentation, following Blaschko lines (the lines of embryologic development of ectoderm).
By infancy, scaling becomes more conspicuous. Blistering occurs less frequently with advancing age, is often focal, and may be caused by secondary staphylococcal infection. Although the disorder is generalized, scaling is particularly verrucous in intertriginous areas and overlying joints. The degree of associated erythroderma and palmoplantar keratoderma is variable. Treatment of this disorder with keratolytics or retinoids is often complicated by the propensity of these agents to increase skin fragility. The mosaic form of epidermolytic hyperkeratosis presents as epidermal nevi with linear streaks of thickening of skin, often with increased pigmentation, following Blaschko lines (the lines of embryologic development of ectoderm).
Lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma are now considered distinct disorders based on clinical characteristics and underlying molecular basis. Both are usually autosomal recessive conditions that almost always present at birth as a collodion baby (Fig. 360-2), a phenotype characterized by taut, shiny skin that has been likened to cellophane (collodion).1,2 This “membrane” leads to eversion of the eyelids (ectropion) and of the lips (eclabium), digital contractures, and, rarely, restricted respiration. In severely affected patients, the cartilaginous portions of the nose and ears may be underdeveloped. Collodion babies should be kept in a moist environment (such as a humidified Isolette), with application of emollients and attention to the increased risks of temperature instability, fluid and electrolyte imbalance (especially hypernatremic dehydration), and infection. The membrane is shed during the first weeks of life. Although most collodion babies eventually adopt the typical characteristics of patients with either lamellar ichthyosis or CIE, some patients show a normal phenotype (lamellar exfoliation of the newborn) or other ichthyosiform disorder.
Within a few months after clearance of the collodion membrane, babies with lamellar ichthyosis show scales that are large, platelike, and hyperpigmented, particularly in patients with darker skin. Underlying erythroderma is minimal, but ectropion and alopecia may be severe. Mutations in the transporter protein ABCA12 may cause lamellar ichthyosis or, when more severe, harlequin ichthyosis. Mutations in genes that encode keratinocyte transglutaminase I, transporter protein ABCA12, FLJ39501, and ichthyin may underlie lamellar ichthyosis. In contrast, infants with nonbullous congenital icthyosiform erythroderma have scales that are lighter in color and finer than those of infants with lamellar ichthyosis. Underlying erythroderma is greater, and alopecia and ectropion may be associated. Not uncommonly, these patients have associated neurologic abnormalities, and this phenotype may be part of other multisystem conditions, such as the neutral lipid storage disease (Chanarin-Dorfman syndrome) or Netherton syndrome (see below). Biopsies show marked acanthosis of the epidermis with a moderately thickened stratum corneum. Several genes may be mutated in this disorder, most commonly involved are the genes encoding 12R lipoxygenase and lipoxygenase 3.
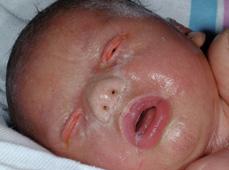
FIGURE 360-2. Collodion baby. Note the taut, shiny skin reminiscent of cellophane, seen with lamellar ichthyosis or nonbullous congenital ichthyosiform erythroderma. (Source: Courtesy of Amy Paller, MD.)
Harlequin ichthyosis is a rare, autosomal recessive trait caused by mutations in the lipid transporter ABCA12 in which affected infants are usually stillborn or die soon after birth from massive, hyperkeratotic plates that obstruct respiratory movements and feeding. Skin tautness and massive plates of scale produce grotesque facial features with severe ectropion and eclabion and mittenlike encasement of fingers and toes. In the rare survivors, the platelike scales are shed post-natally, and an intense exfoliative erythroderma ensues. In general, harlequin babies are treated with supportive therapy, including a humid environment, the aggressive use of emollients, and careful monitoring of fluid and electrolyte needs. Some advocate the use of systemic retinoids for patients who survive the first few weeks of life; it should be recognized, however, that the resultant phenotype with chronic use of retinoids will continue to be that of severe CIE.
Ichthyosis may occur as a component of a multisystemic disease. Netherton syndrome is an autosomal recessive trait caused by a mutation in the SPINK5 gene that encodes the serine proteinase inhibitor lymphoepithelial Kazal-type inhibitor (LEKTI). The syndrome is characterized by the triad of ichthyosis, brittle hair, and an atopic diathesis, often including hypersensitivity to food antigens. Patients usually have generalized exfoliative erythroderma during the neonatal period and early infancy, with failure to thrive, diarrhea, and hypernatremia. Later, an unusual migratory annular pattern of double-edged scaling predominates, called ichthyosis linearis circumflexa. Light microscopic examination of hairs shows a pathognomonic defect with intussusception of the hair shaft, called trichorrhexis invaginata (bamboo hair), which allows the diagnosis to be made.
Patients with trichothiodystrophy, or sulfur-deficient brittle hair, may show a variety of associated defects, including ichthyosis, retardation, and decreased fertility.3 Some patients show photosensitivity, with a severe defect in DNA excision and repair. Under polarizing microscopy, the hair from patients with this autosomal recessive disorder shows a pathognomonic tiger-tail banding pattern.
Sjögren-Larsson syndrome is a rare, autosomal-recessive trait characterized by the triad of ichthyosis, spasticity, and mental retardation. Ichthyosis is usually the presenting sign and patients present with pruritic skin that is thickened and may have a slightly yellow hue. Pathognomonic retinal “glistening dots” are present in most patients by one year of age. Dysplasia of the dental enamel can also be seen. The disorder is caused by a mutation in the ALDH3A2 gene, which causes a deficiency of fatty alcohol oxidoreductase.
Neutral lipid storage disease (Chanarin-Dorfman) is another autosomal-recessive disorder that results from mutation in the gene ABHD5. Patients are most commonly of Mediterranean descent and show a CIE-like ichthyosis. Systemic signs may include neurosensory deafness, cataracts, myopathy, fatty liver, and mild mental and growth retardation. The diagnosis can be confirmed by finding lipid droplets in circulating leukocytes.
Conradi-Hünermann syndrome, or chondrodysplasia punctata, is most commonly an X-linked dominant trait with lethality in males unless in a mosaic form. At birth, patients with Conradi-Hünermann syndrome show bands of ichthyosiform erythroderma that follow lines of Blaschko, reflecting the random activation of the mutant X chromosome. During infancy, these bands resolve, leaving follicular atrophoderma. Systemic signs include focal cataracts and asymmetric limb reduction defects with stippled epiphyses (chondrodysplasia punctata).
CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) is also an X-linked dominant trait caused by a mutation in the NSDHL gene. The ichthyosis and limb defects with epiphyseal stippling are predominantly unilateral.
The triad of progressive scarring keratitis, ichthyosis, and neurosensory deafness characterizes KID syndrome, a rare disorder with dominant and recessive forms that is caused by mutations in genes encoding connexins. Patients often show generalized keratotic plaques, palmoplantar keratoderma, and alopecia.
Darier disease, or keratosis follicularis, is an autosomal dominant trait caused by a mutation in ATP2A2, which encodes SERCA2b, an intracellular calcium pump ATPase. The skin eruption frequently follows intense sun exposure, most commonly in the second decade of life. Greasy keratotic papules show a predilection for seborrheic areas on the face, scalp, neck, upper chest, and back but may occur on extremities or may show a striking photodistribution. Lesions on flexural sites tend to coalesce to form erythematous, eroded plaques. Small keratotic papules and pits are found on palms and soles. Nails are short, fragile, and may show red and white longitudinal streaks as well as v-shaped distal nail nicks. The oral mucosa has a pebbly quality, particularly along gingival margins. The histopathologic appearance of biopsy sections is diagnostic  .
.
PALMOPLANTAR KERATODERMAS
Palmoplantar keratodermas constitute a large group of predominantly autosomal dominant disorders in which the disorder of cornification is largely but not exclusively limited to palms and soles.4,5 The nonepidermolytic and epidermolytic hyperkeratotic types are most common and result from mutations in keratin 1 (and sometimes 6 or 16) and 9 genes, respectively. Hyperkeratosis may be diffuse or limited to papules or even linear bands, as in the striate form of palmoplantar keratoderma. In some forms, particularly Vohwinkel syndrome constriction of the terminal digits may lead to mutilating changes. If keratoses are focal or are accompanied by painful erosions, tyrosinemia type II (Richner-Hanhart syndrome) should be excluded. Other features of this autosomal recessive trait are keratitis and sometimes mental retardation. Dietary restriction of tyrosine and phenylalanine is indicated. The Papillon-Lefèvre syndrome is a recessive trait with diffuse keratoderma and periodontitis leading to premature loss of teeth. In treatment of keratoderma, keratolytics may be used followed by paring. Occasionally, one of the synthetic retinoids (isotretinoin or acitretin) is indicated to preserve function.
DISORDERS OF PIGMENTATION
The genetic basis for albinism in most patients is a mutation either in the gene that codes for tyrosinase, important for melanin synthesis (tyrosinase-negative albinism), or in the gene that codes for P protein (tyrosinase-positive albinism), leading to abnormal transport of melanin to keratinocytes. As a result, children with albinism, an autosomal recessive group of disorders, have decreased pigmentation of the skin, hair, and eyes, along with photophobia, nystagmus, and reduced visual acuity.6,7 Patients with all forms of oculocutaneous albinism must consistently avoid sunlight, use full-spectrum sun protectants, and wear protective clothing (long sleeves, brimmed hat, UV-filtering glasses) to prevent the development of cutaneous malignancy. The National Organization for Albinism and Hypopigmentation (NOAH) has a Web site (http://www.albinism.org) and provides support for families.
The Hermansky-Pudlak syndrome is a rare, autosomal recessive disorder7 characterized by a triad of decreased pigmentation, mild bleeding diathesis, and tissue storage of ceroid material.
Chédiak-Higashi and Griscelli syndromes are autosomal recessive disorders which result from mutations in LYST and myosin Va. The skin and hair assume a pigment-diluted, silvery sheen because of the accumulation of giant melanosomes with inability to transport the melanin granules to epidermal cells. Both disorders may show evidence of immunodeficiency, with the development of an “accelerated phase” by early childhood, which leads to the proliferation of atypical lymphocytes and histiocytes, multiorgan infiltration, and pancytopenia. Without successful bone marrow transplantation, most patients with Chédiak-Higashi or Griscelli syndrome die during this accelerated phase. Patients may develop neurologic deficits, particularly in Chédiak-Higashi syndrome and in type I Gris-celli syndrome. Patients with type II Griscelli syndrome (caused by mutations in Rab27a) are at particularly high risk for hemophagocytic lymphohistiocytosis.
Children with piebaldism display discrete patches of depigmentation involving the central forehead, anterior trunk, and midportions of the upper and lower extremities.7 A midline white forelock is characteristic. Absent or grossly abnormal melanocytes are seen in the involved skin. The condition is inherited as an autosomal dominant trait owing to mutations in the c-kit protooncogene, which directs melanoblast proliferation, migration, and differentiation.
Waardenburg syndrome is also an autosomal dominant disorder associated with mutations in PAX 3, with depigmentation of hair, skin, and irides. Its phenotypic manifestations are most commonly lateral displacement of the inner canthi (dystopia canthorum), broad nasal root, and confluent eyebrows. Patients without the facial features may have mutations in the MITF gene (type 2), and patients with associated Hirschsprung disease (type 4) may have autosomal recessive mutations in 1 of 4 other genes encoding endothelin, endothelin receptor, Slug, or Sox 10. Sensorineural hearing loss occurs overall in 20% of patients and indicates the critical role of pigment in directing development of hearing.
TUMOR SYNDROMES
Pediatric patients with the autosomal dominant type I neurofibromatosis (von Recklinghausen disease) usually show only multiple café au lait spots on examination.8-11 Six or more of these evenly brown-colored, sharply defined macules are present in more than 99% of patients. Additional café au lait spots may continue to appear during the first 5 years of life but by definition must be larger than 0.5 cm in diameter in the prepubertal child (1.5 cm in a postpubertal individual) to be counted for diagnosis. Definitive diagnosis requires the presence of other features, such as axillary freckling (Fig. 360-3); neurofibromatosis in a first-degree relative; pseudoarthrosis; plexi-form neurofibroma; sphenoid dysplasia; optic glioma or Lisch nodules (dome-shaped hyperpigmented iris hamartomas that are multiple, bilateral, and present in the majority of patients of school age and older). Two interacting genes, tuberin or hamartin, may be mutated to cause this disorder.
The plexiform neurofibromas often manifest during infancy and early childhood as a large café au lait spot, often irregular, with associated hypertrichosis. They can be deeply invasive, grow progressively, and be difficult to eradicate. Dermal neurofibromas present as soft sessile or pedunculated nodules, usually first apparent during puberty. Optic gliomas occur in 15% of patients but are not treated unless they are actively growing or causing clinical problems. Defective tumor suppression increases the risk of malignancy; central nervous system tumors and nonlymphocytic leukemias may appear during childhood and neurofibrosarcomas and malignant schwannomas after 20 years of age. Perhaps the most important reason for early diagnosis is the awareness that speech and learning disabilities occur in almost a third of patients. 
Hypopigmented macules ranging from 1 to 4 cm in diameter are the most consistent cutaneous finding in tuberous sclerosis, an autosomal dominant neurocutaneous disorder.9,10 Macules are sometimes shaped like ash leaves (ash-leaf spots), but they may be round, oval, irregular, or confetti-like in configuration. The presence at birth of connective-tissue nevi (slightly elevated flesh-colored plaques with a rough or leathery surface, particularly on the forehead [fibrous forehead plaque]) may allow early definitive diagnosis. The plaques of collagenomas that occur in tuberous sclerosis are also known as “shagreen patches.” Numerous skin-colored to vascularized papules (adenoma sebaceum) appear on the central face during childhood in most patients with tuberous sclerosis (Fig. 360-4). Periungual, subungual, and gingival fibromas are not usually seen until puberty or adulthood. The majority of patients with tuberous sclerosis have seizures, often with onset by infancy, but 40% have normal intelligence. Tumors of the central nervous system, kidneys (angiomyolipomas), heart (rhabdomyomas), and eyes (retinal astrocytic hamartomas/phakomas) occur most commonly. The Tuberous Sclerosis Alliance (http://www.tsalliance.org) provides support to families.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


